Pathophysiology of Vascular Manifestations in Systemic Lupus Erythematosus
Introduction
Systemic Lupus Erythematosus (SLE) is a potentially fatal, chronic, multisystem autoimmune disorder. As defects can occur in various parts of the immune cascade, clinical presentations may vary significantly [1]. These clinical features range from musculoskeletal to neuropsychiatric, renal, cutaneous, gastrointestinal, pulmonary, cardiac, vascular, haematological, ophthalmological and immunological manifestations [2]. In a prospective, Europewide, decade-long study, the most common symptoms of SLE were arthritis, malar rash, nephropathy, photosensitivity, neurologic symptoms, fever, Raynaud’s phenomenon, serositis, thrombocytopenia and oral ulcers [3]. Notably, coronary heart disease, atherosclerosis and thrombosis are common in SLE [4,5], and small- and medium-vessel vasculitis across affected organ systems prevail in up to 36 % of SLE cases [6]. Adding to the complexity of the disease are varying laboratory abnormalities‚ from hematological and serological changes to negative serology or the occurrence of specific autoantibodies [1]. SLE predominantly affects women between puberty and menopause, which suggests an endocrine influence on disease onset and progression. This is underpinned by the fact that the female-to-male ratio in children is rather low and increases significantly in childbearing age [7].
Apart from hormonal factors, genetic, ethnic, environmental, psychological and socioeconomic influences have also been identified [7,8]. Due to this etiologic diversity, a considerable variation in incidence and prevalence rates among different countries is to be expected. Indeed, North America has the highest incidence and prevalence rates, while African populations show the lowest incidence rates, and Australia has the lowest prevalence. In Europe, incidence rates ranged between 1 and 4.9 per 100.000 person years, and prevalence settled between 16.2 and 110 cases per 100.000 [9]. According to estimates, around 4000 SLE patients can be expected among the Austrian population [10].
While the 5- and 10-year survival rates in SLE improved significantly in the second half of the 20th century (from 75 % to 95 % and from 63 % to 91 %, respectively), this improvement has slowed down from the 1980s onwards, despite further advances in diagnostics and treatment [11]. As direct mortality decreased, long-term complications and comorbidities might be of increased interest. This conclusion is supported by the fact that SLE is generally associated with an increased risk of premature death [1]. Traditionally, SLE has been viewed to have a bimodal pattern of mortality, presenting with peaks of early mortality (<1 year after onset) due to disease activity or infection, and of late mortality (∅ 8.6 years after disease onset) due to cardiovascular events [12]. A substantial number of instruments have been developed in order to facilitate diagnosis and classification of SLE. Diagnosis of SLE has relied on the American College of Rheumatology (ACR) criteria[13] for years but has been challenged by alternative classification concepts since. Earlier this decade, the Systemic Lupus International Collaborating Clinics (SLICC) criteria were introduced and were proven to be equally specific, but more sensitive [14].
Most recently, an initiative for new European League Against Rheumatism EULAR/ACR criteria exhibited promising results in adult-onset SLE (aSLE) [15] but did not yield superior global results in a retrospective study of pediatric-onset (pSLE) cases [16]. Apart from diagnostic criteria, many different disease activity indices have been developed in order to quantify clinical outcomes, compare patient groups cross-sectionally, and measure disease activity longitudinally [17]. Among various scoring systems, the SLE Disease Activity Index 2000 (SLEDAI-2k) is a commonly used index which allows for the prediction of global disease activity and the occurrence of new disease flares [18]. In contrast to the aforementioned tools measuring SLE disease activity, the SLICC/ ACR Damage Index (SDI) assesses irreversible damage to 12 organ systems in SLE patients regardless of their cause, including various organ manifestations related to vascular morbidity [19].
For the onset and progression of SLE, multiple risk factors have been identified. Among the most prominent candidates, tobacco smoking, environmental exposure, gender influences and genetic risk alleles have been proposed [20-22]. With respect to current or recent tobacco smoking, it has been shown to be strongly associated to anti-double-stranded DNA (anti dsDNA) seropositivity and the development of SLE, even though the underlying mechanisms have not yet been completely understood [20]. Occupational exposure to crystalline silica dust has also been demonstrated to strongly correlate with SLE prevalence, as it is believed to increase apoptotic material in a pro-inflammatory environment [23]. In women, estrogen-related exposures (e.g. early age at menarche, oral contraception etc.) were identified to be associated with SLE susceptibility, and explained by immunomodulatory functions of sex steroid hormones [24]. Moreover, a multitude of genetic risk alleles have been identified to contribute to certain SLE subphenotypes, leading to the establishment of a genetic risk score (GRS) for SLE [25].
SLE and Cardiovascular Disease
Patients suffering from SLE are at risk for premature cardiovascular disease (CVD). Apart from manifestations such as venous thromboembolism, Raynaud’s phenomenon, vasculitis, peri-, myo- and endocarditis, valvular disease and aneurysm, most research has been conducted on atherosclerosis and arterial thromboembolic events. Prevalence of atherosclerosis has been confirmed to be significantly elevated in SLE patients in metaanalyses of studies providing data of surrogate markers, namely carotid intima-media thickness and prevalence of carotid plaques [26]. Strikingly, CVD accounts for more than one-third of all deaths among SLE patients [27]. In a prospective cohort study among 119,332 women, the relative risk of a cardiovascular event (myocardial infarction, stroke, coronary artery bypass grafting, angioplasty) in SLE patients was 2.26 after multivariate adjustment for confounding factors, among which “traditional” cardiovascular risk factors [28]. In some studies, up to 50-fold incidence rate increases have been identified for myocardial infarction in SLE patients, e.g. in young women of reproductive age [29].
Both adult- and pediatric-onset SLE have been identified as independent risk factors for premature atherosclerosis [30], coronary heart disease [31], myocardial infarction [32] and/ or death in young premenopausal women [33,34]. Reciprocally, many “traditional” cardiovascular risk factors, such as metabolic syndrome, smoking and hyperhomocysteinaemia are more prevalent among SLE patients [4]. Altered severity of these risk factors could, however, not be demonstrated to independently account for the significantly increased risk for adverse events (coronary heart disease and stroke) in SLE [31]. Additionally, lupus nephritis (LN), one of the most common organ manifestations in SLE, increases odds to develop atherosclerotic plaques by more than two-fold compared to non-LN patients and healthy controls [35]. Apart from atherosclerosis and its associated morbidities in the arterial branch of the vascular system, venous thrombotic events occur in SLE at elevated rates: Different studies identified a 20-year risk of around 10 % for both deep venous thrombosis and pulmonary embolism [36].
Up to two-thirds of SLE patients present with Raynaud’s phenomenon at a given point in time [37], but prevalence differs significantly between sources [38], with one prospective study reporting an incidence rate of around 16 percent during a 10- year period [39]. Among SLE patients, vasculitis presents as a heterogenous class of disease which rarely affects large [40] but more commonly medium or small vessels and can be divided into primary and secondary forms [41]. Vasculitis afflicts different organ systems, which explains a vast array of possible, heterogenic symptoms due to cutaneous, musculoskeletal, mesenteric, hepatic, pancreatic, coronary, pulmonary, retinal, neurologic and renal manifestations [6]. As the most common, strictly cardiac manifestation of SLE, around a quarter of all SLE patients develop pericarditis, with subclinical disease being even more prevalent [42]. In these patients, pericardial effusion, cardiac tamponade, pleuritis and myocarditis occur more frequently [43,44]. Valvular abnormalities, such as valvular thickening, fibrosis or regurgitation, are observed in up to 18 % of SLE patients [45]. A subset thereof are cases of Libman-Sacks, or “atypical verrucous” endocarditis, which most commonly affects the mitral valve, but is typically clinically silent and does infrequently lead to complications [42,46].
Rarely, namely in under 10 % of cases, SLE patients develop clinically overt myocarditis, but subclinical involvement might be more frequent [44,46]. Myocarditis may facilitate the development of nonspecific electrocardiographic (ECG) changes, heart block, arrhythmia, tachycardia, heart failure and cardiomyopathy [44,47]. Whether the latter is primary or secondary in SLE patients is not easily distinguishable, as signs and symptoms are similar regardless of its cause [46]. It is, however, likely that most cardiomyopathies in SLE are secondary and emerge due to coexisting hypertension, coronary artery disease and atherosclerosis, small vessel disease, cardiac microcirculation thrombosis, renal failure, valvular disease or drug toxicity [42,46]. Rates of different conduction tissue abnormalities in SLE diverge widely. While peri- and myocarditis frequently present with tachycardia, atrioventricular and branch blocks occur rarely in adults [42,46]. In infants from anti-Ro and anti-La autoantibody-positive mothers, cross-placental Immunoglobulin (Ig) G transmission can cause transient neonatal SLE syndrome [48]. This condition often involves isolated, first to third degree, permanent heart block with onset in the second or third trimester, which presents with fetal bradycardia, and, in some cases, results in cardiomyopathy [49-51].
In adult-onset SLE patients, numerous cases of aneurysm or dissection of variable severity and of different vessels have been reported with onset at strikingly young ages, among which thoracic [52-54,54] and abdominal aortic [54-56], coronary [57,58], pulmonary [59], carotid [60], basilar [61], cerebral [62,63], hepatic [64] and mesenteric [65] artery aneurysms. Some cases have been reported as early as in childhood [66] and adolescence [67,68]. Generally, reports on aneurysm in SLE are less common than literature on other cardiovascular complications such as pericarditis or atherosclerosis, but some observations have been made. Regarding coronary artery aneurysm in SLE patients, an odds ratio (OR) of 4.09, adjusted for demographics and related risk factors, but excluding traditional CVD risk factors has been described in a retrospective, population-based case-control study [69]. With respect to aortic aneurysm (AA), a similar study has described a non-adjusted OR of 4.5 [70]. In another populationbased retrospective study, incidence rate ratios (IRRs) of 2.63 for AA and 5.7 for aortic dissection were detected along with correlating traditional and non-traditional risk factors, such as SLE disease duration > 3 years and end-stage renal disease [71]. In absolute numbers, AA does not occur commonly in SLE patients, but when it does, it has a substantial negative influence on patient survival [72]. Apart from the higher incidence and mortality of aneurysm in SLE patient’s vs the general population, the age of onset is significantly lower among the former: In two meta-analyses, the median age for onset of AA was around 45 years [72,73], which is around 20 years younger compared to the general population [74]. In addition to that, patients with SLE complicated by abdominal AA are struck with disease around 30 years earlier [56].
Apart from the aforementioned manifestations, antiphospholipid syndrome (APS) is a separate, but often accompanying disease entity to SLE. APS regularly causes thromboembolic events and other major cardiovascular complications, including stroke, deep vein thrombosis, pulmonary embolism and valvular disease [75].
Pediatric-Onset SLE
Incidence and prevalence rates in pediatric-onset SLE are substantially lower than in its adult-onset counterpart: Only 15-20 % of cases of SLE present before the age of 18 years [34] and a mere 8 % develop the disease before the age of 14 years [3], which is why many studies report data from adult populations only [1]. In studies from Europe and North America, the annual incidence rate of SLE in children and adolescents under 16 years of age was less than 1 in 100,000 persons [7,76]. According to a study conducted in the late 1990s, the mean annual incidence rate for SLE in patients under 16 years of age was 0.4 per 100,000 person years across three eastern Austrian regions [76]. Patient survival rates are similar between adult- and pediatric-onset SLE, however, SLErelated deaths are more common in pSLE due to a generally lower baseline mortality in children [33]. This can be explained by the fact that, compared with adult-onset SLE, patients with pSLE tend to undergo a more aggressive clinical course (expressed in higher mean SLEDAI scores), a higher rate of major organ involvement, exposure to tissue damage at a younger age, longer overall disease duration, and an increased risk of developing vasculitis [32,33,77]. As a result, long-term-morbidities play an even more prominent role in pSLE patients [33]. While initial diagnosis at a relatively high age can be associated with a lower risk of long-term morbidity [28], delay in diagnosis of SLE is associated with increased damage to vital organ systems [78]. As cumulative organ damage as per SDI value has been proven to, inter alia, significantly increase with disease duration in childhood-onset SLE [79], this even more so underlines the fact that early diagnosis of SLE is important to prevent major organ damage [80] and to lower the rate of disease flares [81,82].
With longer disease duration, endothelial dysfunction (and therefore early-stage atherosclerosis) progresses over time in both pSLE and aSLE patients [83–85]. As a precursor to morphologic changes of the arterial wall, endothelial dysfunction decreases along with flow‐mediated dilation (FMD), a sonographically determinable surrogate marker which corresponds to the endothelium’s capacity to mediate reactive hyperemia [86]. Moreover, increased carotid artery stiffness, also a suspected precursor to atherosclerosis, has been demonstrated in patients suffering from both active and inactive pSLE, even in those with relatively short disease duration and good disease control [87,88]. In younger pSLE patients, arterial stiffness and consecutive morphologic vascular changes have been shown to correlate with disease activity, but not (yet) with traditional CVD risk factors [89]. Other data suggests that carotid intima media thickness (CIMT), a surrogate marker for atherosclerosis, does, however, correlate with both traditional (age, body mass index, male gender) and nontraditional (Azathioprine, low- and high-, but not medium-dose glucocorticoid treatment) risk factors in pSLE patients (Figure 1) [90]. As disease progresses, reducing the cardiovascular risk for patients suffering from pSLE by optimal control of disease activity and reducing the “add-on” of traditional cardiovascular disease (CVD) risk factors is, by all means, of great importance [26,33,91,92].
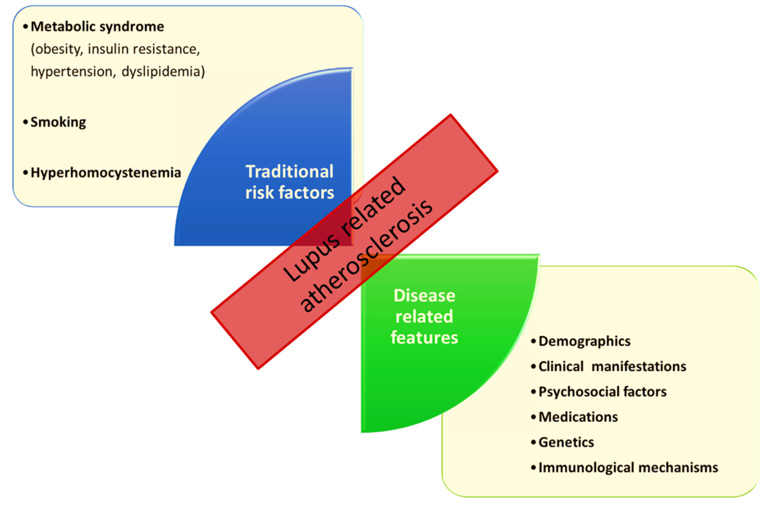
Figure 1: Traditional risk factors and disease-related features in lupus-related atherosclerosis (4)
Pathophysiology
SLE is a chronic autoimmune disease which encompasses many landmark mechanisms of autoimmunity, such as loss of tolerance against autoantigens, lymphoproliferation, polyclonal autoantibody production, immune complex disease (Arthus/type III hypersensitivity reaction) and target organ inflammation [93,94]. On its most basic level, it is caused by an inappropriate immune response against self-antigen, particularly nucleic acid-containing particles. Because several intensively intertwined processes and aberrations interplay in SLE, the origin of its autoimmunity has not been conclusively identified to date. In a highly simplified model of its disease mechanisms, impairments of (apoptotic, necrotic) cellular remnant clearance could be chosen as an arbitrary starting point. From there on, the accruing autoantigens are opsonized by naturally occurring autoantibodies (NAbs) before being targeted by the complement system and phagocytes. Now, many different pathways commence: Local inflammatory reaction by cells of the innate immune system and complement complexes leads to early signs of tissue inflammation, cytokine release, and attraction of additional leukocytes. Antigen-presenting cells (APCs) migrate to lymphatic organs, where they are sampled by lymphocytes in order to establish highly specific cellular and humoral immunization. Developing specificity of immune reactions and rising numbers of autoantigen-specific antibodies perpetuate and extend autoinflammation which peak in vicious disease flares resulting in long-term target organ damage [1,95,96].
As Figure 2 suggests, susceptibility for developing autoimmunity has been associated with a plethora of candidate genes regulating the immune response in various stages. Genetic variants can negatively impact self-tolerance and are typically labelled as lossor gain-of-function mutations. For instance, a loss-of-function in gene motifs coding for parts of the physiological clearance of cell remnants might affect the development of SLE similarly to how a gain-of-function mutation of pro-inflammatory T cell signaling would and vice versa. Some severe cases of SLE present with early onset, which is suspicious for monogenic SLE, a concept of disease entities induced by solitary mutations in single genes [93,97,98]. Apart from these rare and mostly pediatric-onset cases of SLE, multiple hits of different flavors accumulate before a “susceptible” genetic predisposition lapse into active disease Figure 3.
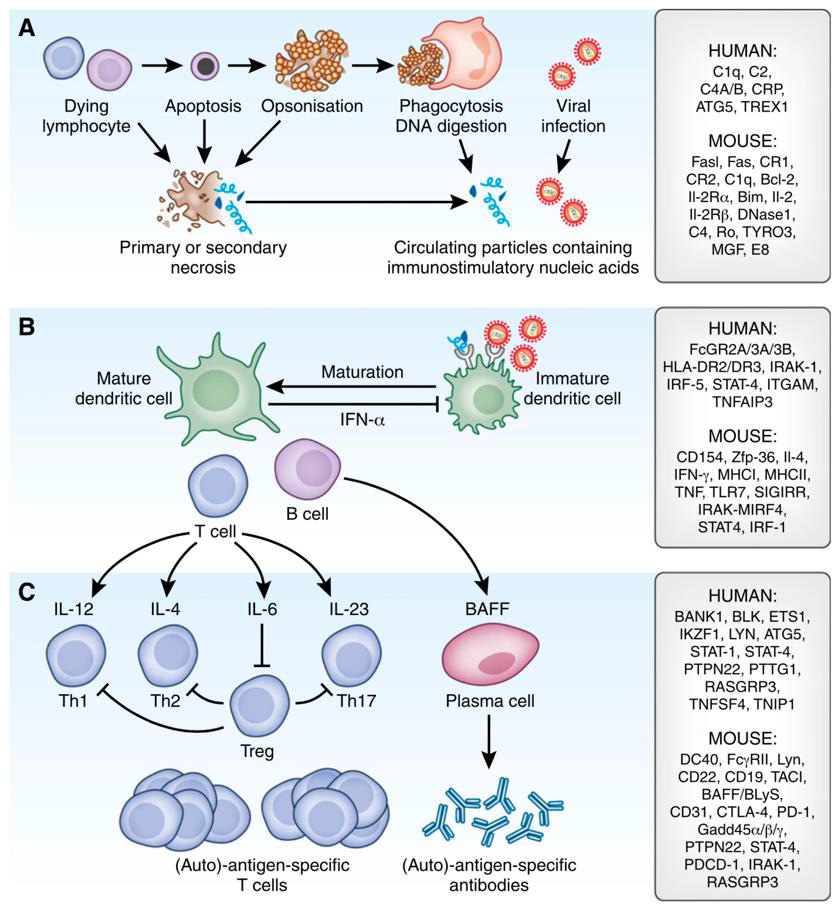
Figure 2: Pathomechanisms of SLE and a selection of candidate genes (93).
These dysregulated autoimmune processes involve both the innate as well as the adaptive immune system, which explains why certain types of antibodies are commonly found in SLE. Foremost, antinuclear antibodies (ANAs) are identified in almost all cases of SLE [99]. As members of the ANA specifity, anti-Smith (Sm) and anti-dsDNA antibodies are two examples of commonly found antibodies in SLE [1].
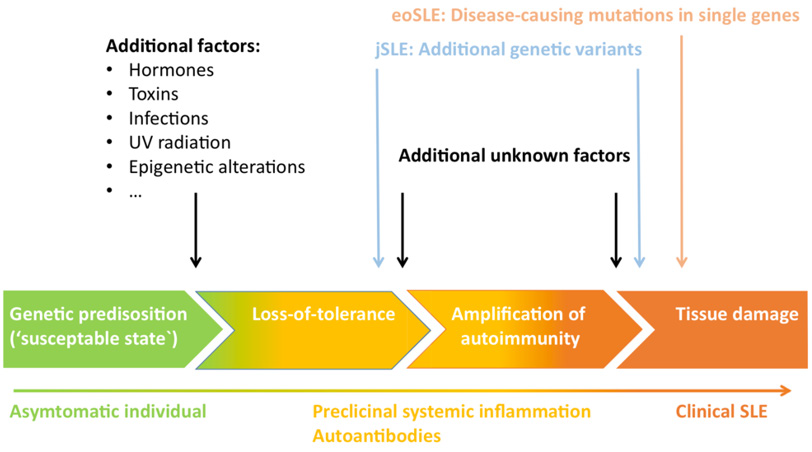
Figure 3: Timeline of SLE disease progression and pathogenic hits (98)
Cell Death, Nuclear Remnants and Clearance
In order to develop immunization against self-antigens, nuclear epitopes have to be accessible to antigen-presenting cells (APCs). In physiologic homeostasis, the overbearing presentation of self-antigens is avoided by rapid cell death clearance, which is a complex process involving components of the complement system, dendritic cells (DCs) and phagocytes, as well as a multitude of other factors such as antibodies and other proteins, enzymes, cytokines and opsonins [100]. Due to unfortunate immunoregulatory defects in SLE, low levels of chromatin in extracellular compartments (e.g., after necrosis, impaired apoptosis or NETosis, see below) cannot be maintained. Consequently, protein antigens and immunostimulatory nucleic acids (acting as immune adjuvants) are targeted and presented by plasmacytoid dendritic cells (pDCs), which serve as a link between the innate and adaptive immune system [93,101]. In other words: If cell debris is not efficiently cleared, a first step towards an excessive immune response is triggered. Apart from extracellular cell debris due to impaired clearance of dysfunctional cells, SLE autoantigens can also present on the surface of apoptotic cells [102]. With regard to reasons for cell death, external noxious influences such as UV radiation of the skin is hypothesized to contribute to apoptosis, secondary necrosis or impaired clearance, but this has recently been subject to discussion [103,104].
Among many different hypothesized dysfunctional factors, complement component (C) 1q deficiency is believed to add to the breakdown of self-tolerance [102]. This shortage might be traced back to impaired local synthesis of C1q in its main producers, namely macrophages (Mφ) and dendritic cells [105]. To further complicate the involvement of phagocytes, functional Mφ/ monocyte (Mo) defects have been identified to contribute to SLE, including decreased phagocytosis, antigen presentation (MHC-II/ HLA-DR, CD80), Interleukin (IL)-1b production, autologous mixed lymphocyte reaction, and T-cell activation [106]. Apart from these defects, increases in pro-inflammatory M1 and M2b, and decreases in anti-inflammatory M2a and M2b subsets skews Mφ populations towards a more autoinflammation-prone profile [107]. Additional mechanisms, such as the inhibition of class A scavenger receptors by autoantibodies, are also believed to impair phagocyte function [108]. Aside from a reduction in cell and immune complex clearance, these functional defects lead to the impaired (co)stimulation of lymphocytes and the accumulation of apoptotic material and immune complexes in target organs (see below).
Innate Immunity
In general, nucleic acid-containing immune complexes and cytoplasmatic RNA and DNA are known to be potential stimuli for the activation of the innate immune system [1]. In SLE, type I interferons (IFNs) are produced in a response mainly mediated via endosomal toll-like receptors 7 and 9 (TLRs) and TLR-independent nucleic acid sensors. Apart from migrating pDCs, which are the major source of IFNα, a variety of cells participate in the production of pathogenic type I-IFNs, among which fibroblasts, epithelial cells and bone-marrow resident neutrophils [109,110]. Belonging to the group of pattern recognition receptors (PRRs), TLRs are membrane receptors located either on the cell surface or intracellularly in the endosomal-lysosomal compartment, notably in dendritic cells (DCs) [111]. Because of their function to bind pathogen-associated molecular patterns (PAMPs), TLRs are generally considered to contribute to antiviral immunity [112] in a host of different cell types. As opposed to myeloid dendritic cells (mDCs), which express functional TLR7 but not TLR9, pDCs express both of these subtypes [113]. Depending on their variant, TLRs recognize different molecular patterns. In particular, TLR7 binds to single-stranded RNA (ssRNA) and imidazoquinoline compounds [114,115], and TLR9 binds unmethylated cytosine-guanine dinucleotide (CpG)-rich DNA [116]. Additionally, TLR7 and TLR9 can be activated by immune complexes internalized into the cytoplasm via the interaction of Fc γ receptor II (FcγRII) and cell fragments [114,116]. Ultimately, TLRs couple nucleic acid detection to essential immune functions, such as the induction of cytokine responses, the presentation of antigen to lymphocytes, and the enhancement of virus-specific antibody responses [117].
Data from murine models suggests potentially pathologic functions of TLRs in SLE (Figure 4). With regard to the production of autoantibodies, TLR7 and TLR9 exert analogous functions – the former propagating anti-RNA (e.g. anti-Sm), the latter facilitating anti-DNA production. Although both TLRs are associated with components of the same downstream signaling pathways, TLR7- deficiency was linked to decreased lymphocyte and pDC activation, as well as ameliorated end-organ (e.g., renal) morbidity. Moreover, TLR9 has been described to downregulate the expression of IFNα [118]. However, in the presence of DNA-containing immune complexes, IFNα seems to be upregulated in a TLR9-dependent mechanism [119]. A similar effect is attributed to TLR7-dependent anti-RNA autoantibodies, which are believed to also enhance the production of type I IFNs. In this respect, opposing roles are attributed to TLR7 and TLR9 in the pathogenesis of SLE [118]. Also, some observations attest a strongly correlated pathogenic function of autologous RNA-containing immune complexes in conjunction with TLR7 in innate immune activation, IFN production and the development of SLE [120].
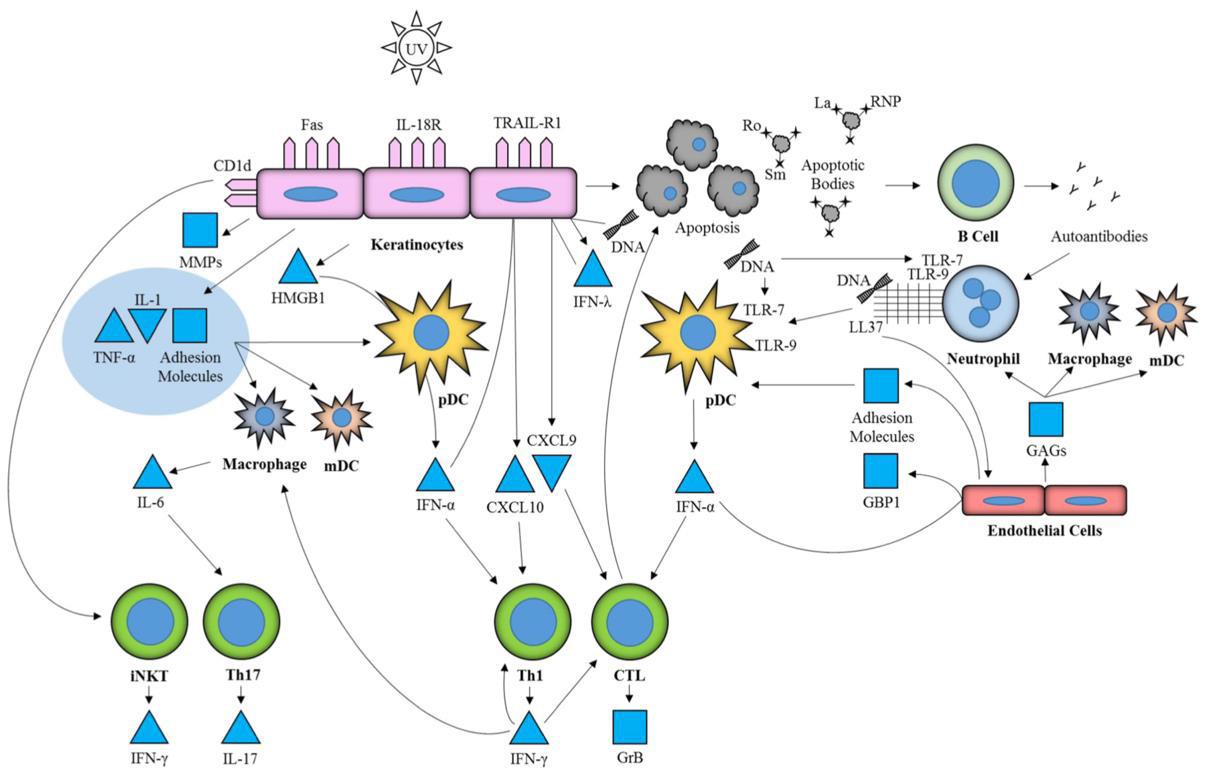
Figure 4: Involvement of TLR7 and 9 in cutaneous lupus erythematosus (103)
Other data suggests that TLR-independent innate immune pathways might also contribute to SLE pathogenesis in cells of the innate immune system: RNA helicases, such as RIG-like Receptors (e.g. RIG-I, MDA5), can bind and destabilize distinct (viral) RNA patterns, and form a signaling complex leading to IFN and IFNstimulated gene expression and the production of type I IFNs and pro-inflammatory cytokines via mitochondrial antiviral-signaling protein (MAVS), nuclear factor-κB (NF-κB), interferon regulatory factor (IRF) 3 and IRF7 (112,121). With regard to DNA recognition, a TLR9-independent mechanism was identified, which can be, for instance, relevant in TLR9-lacking mDCs: In a nutshell, the inhibition of Cyclic GMP-AMP synthase (cGAS) significantly blunted autoimmunity, particularly the expression of IFN-induced genes and inflammatory cytokines, in two autoimmunity-prone murine models (Trex1-/- and DNaseII-/-) [122]. In about two-thirds of adult-onset SLE patients and virtually all pediatric-onset SLE patients, overstimulation of type I IFN-producing cells and vastly elevated levels of IFN-dependent downstream products suggest that type I IFNs are pivotal contributors in the pathogenesis of SLE [123]. These type I IFNs cause the expression of a measurable “IFN signature”, consisting of multiple type I IFN-inducible genes expressed primarily in peripheral blood mononuclear cells (PBMCs) [124,125]. It can be used as a biomarker of disease activity and might be useful in selecting individual treatment regimens [126]. Due to the significance of IFN-I as a primary pathogenic factor in SLE, gain-of-function mutations in the type I IFN pathway are assumed to associate with disease onset and severity [112].
Interferons have many pro-inflammatory key roles in SLE, which overlap among many species of type I-III IFNs, and target cells of both the innate and adaptive immune system. Most notably, IFNα stimulates monocytes and their development into myeloid dendritic cells (mDCs), as well as T-helper (TH) 1 and cytotoxic T CD8+ cells. B cell differentiation, autoantibody production and class switching, natural killer (NK) cell cytotoxicity and NETosis (neutrophil extracellular trap formation) are also facilitated. Moreover, IFNα suppresses the development of TH2, TH17 and anti-inflammatory regulatory T (Treg) cells, is believed to trigger apoptotic death of certain tissue cells, and to impede clearance of nuclear antigens by macrophages [110,127]. Therefore, type I IFNs facilitate pathognomonic processes in lupus, such as an increase in autoantigen loads, the augmentation and presentation of autoantigens via APCs, the production of IgG autoantibodies and inhibition of immunosuppressive activity [127]. In this respect, it is not surprising that type I IFNs are reasonably hypothesized to promote premature cardiovascular disease in SLE [128]. Overall, immune alterations observed in SLE resemble those characteristic of chronic viral infection [93,112,123], such as: Sustained expression of type I IFNs; increased and sustained production of proinflammatory mediators such as IL-6 and 1IL-10 as well as tumor necrosis factor (TNF); altered expression of cell surface receptors including programmed death ligand 1 (PD-L1) and TNF-related apoptosis-inducing ligand (TRAIL, TNFSF10); and a shift in T cell differentiation towards a follicular T helper cell phenotype. These alterations induce, among others, sustained and poorly regulated macrophage activation, impaired T cell function and regulation of cell death, excessive B cell differentiation, the production of autoantibodies and immune complexes and widespread tissue and organ inflammation and damage [1].
In this regard, Epstein–Barr virus (EBV) infection has been highlighted as a potential trigger of the disease. Precisely, defective CD8+ T cell response to EBV infection might lead to an increase in intra- and extracellular EBV DNA, which would induce a shift toward a CD4+ T cell mediated immune reaction, and therefore lead to an increase in the production of autoantibodies (129). Aptly, specific antibodies to EBV nuclear antigens (e.g. EBNA-1) have been demonstrated to effectively cross-react with autologous dsDNA [130]. As opposed to mechanisms linked to viral infection, primarily antimicrobial mechanisms of the innate immune system have recently received attention in the pathogenesis of adultand pediatric-onset SLE, namely cell death and the formation of neutrophil extracellular traps (NETs), most likely after exposure to anti-RNA antibodies [131]. In NETosis, neutrophils actively release chromatin (and therefore autoantigens such as dsDNA), bactericidal proteins and enzymes, and immunostimulatory molecules (e.g. IL-17) into the extracellular space in order to fend off microbial infection [132]. Therefore, NETting neutrophils are believed to, among others, contribute to the activation of innate immunity, the characteristic stimulation of IFNα production of pDCs, and target organ damage [132,133]. A distinct neutrophil subset, namely lowdensity granulocytes (LDGs) are prone to form NETs, which might be due to an increased sensitivity to IFNα stimulation [131]. This subclass is of great interest in SLE, as it has been linked to skin damage, vasculitis, and endothelial cell death [132].
Apart from a potential role of naturally occurring antibodies, the complement system is the main participant of the humoral innate immune system involved in the pathogenesis of SLE [134]. Under physiological conditions, complement has an independent role in cytolysis via its soluble or membrane-bound membrane attack complex. Three (classical, lectin, and alternative) pathways start the complement cascade either after opsonization of immune complexes (via C1q), ligation of recognition molecules (such as mannose-binding lectin, or MBL), or, in case of the alternative pathway, spontaneously (Figure 5). In the further course of the complement cascade, proinflammatory recruitment of leukocytes (through C3a, C5a), and an increase in specific autoantibody production (C3d) facilitate inflammation. Additionally, opsonization (C1q, C3b, C4b) leads to immune complex and cell debris clearance by phagocytes (100,102). In SLE, C1q suppresses the production of SLE immune complex-induced IFNα by directing stimulatory immune complexes to monocytes rather than to IFNα- producing pDCs, when both of these possible targets are present [135]. Accordingly, C1q and a number of additional complement components are decimated in SLE patients by autoantibodies, most frequently by anti-C1q, anti-MBL and anti-C1s autoantibodies [136]. While C1q physiologically attaches to the Fc domains of IgG and IgM antibodies as an entry point into the classical complement pathway, only immune complexes containing both anti-C1q and C1q seem to be correlated with tissue inflammation in SLE, and likely amplify complement activation rather than inhibition [102,136,137].
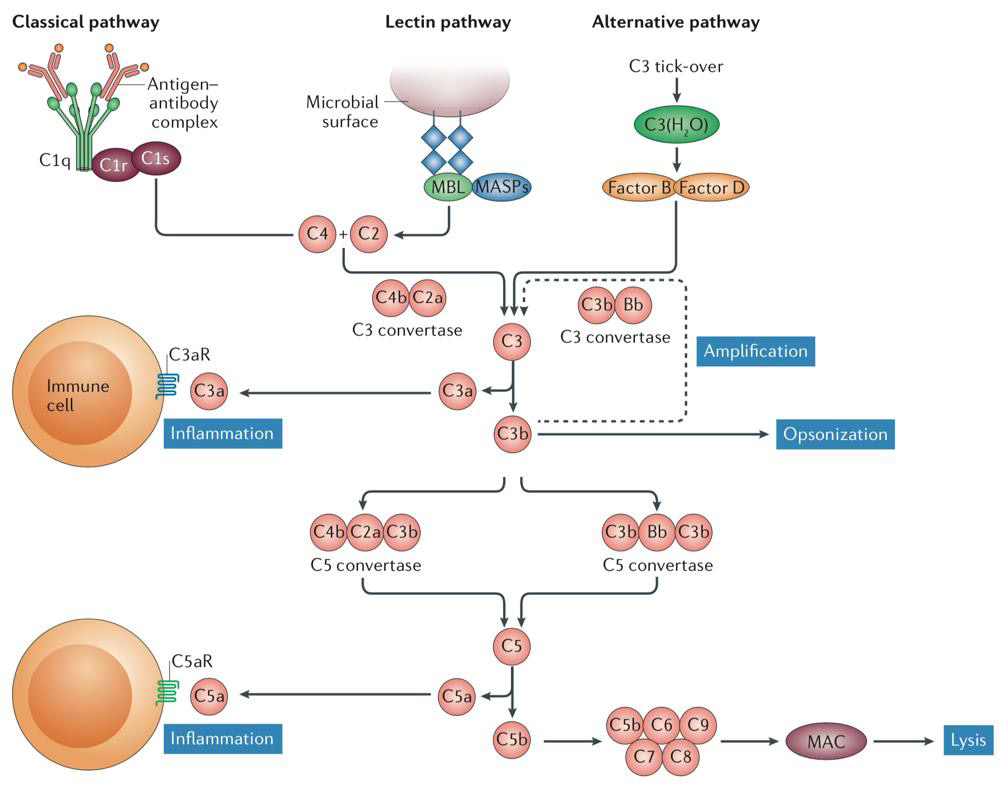
Figure 5: Overview of the complement cascade and its three main pathways (102).
Accordingly, congenital C1q deficiency is highly associated with SLE [103], and defects of C1q production in Mφ and DCs are highly suspect [105]. In conjunction, these findings strongly imply that C1q exerts a protective role in SLE. In contrast to the loss of C1q, other complement components are also diminished in patients’ blood samples. This circulatory complement depletion most commonly manifests as a reduction in C3 and C4, as these components accumulate in inflamed tissues (see below) [102]. While data regarding the impact of anti-MBL activity on disease progression is inconsistent [136,138,139], anti-C1s engages C1s’ proteolytic activity and is therefore believed to be partially responsible for the reduced levels of C4 observed in SLE [136,140], but C1s deficiency was shown not to independently induce SLE [105]. In lupus-prone mice, downstream inhibition of immune cell complement receptors (C3aR, C5aR) is beneficial and reduces symptoms, but a general C3-deficiency is associated with enhanced symptoms [102]. This raises the question whether complement-induced lysis might be beneficial in SLE, while complement-mediated inflammatory response has adverse effects.
Adaptive Immunity
Cells of the adaptive immune system are essential immune mediators in the pathogenesis of SLE as they exert a number of functions, predominantly the production of pathogenic autoantibodies (Figure 6). Beyond the build-up of the pathognomonic humoral adaptive (auto-)immunity, lymphocytes reproduce, adapt, and stimulate immune cells in order to develop, enhance and perpetuate autoimmunity and autoinflammation [95,141]. Considering the previous remarks on the excessive activation of innate immunity in SLE, lymphocyte tolerance to autoantigens is constantly challenged by downstream stimulation. As soon as self-tolerance is lost due to distinct aberrations, autoreactive cells enhance autoimmunity beyond its expected magnitude from innate immunity alone [142]. Thymocytes (T cells) and bursa of Fabricius cells (B cells) are primary drivers of disease chronification, because subsets of these lymphocytes, namely memory T and B cells as well as plasma cells, memorize prior immunization against SLE autoantigens [93]. T cells in SLE deviate from those of healthy individuals in a number of – sometimes contradictory – ways. The T cell phenotype in SLE is distinctly different than in healthy individuals: In SLE, a hyperactive T cell population reacts to low thresholds for activation by secreting pro-inflammatory cytokines, cooperating with B cells, stimulating autoantibody production, and accumulating autoreactive T cells [143,144]. Even though generalized lymphocytopenia is common in SLE, specific T cell populations either proliferate or decline [1]. When considering certain individual thymocyte subgroups, apparent deviations can be discerned in SLE:
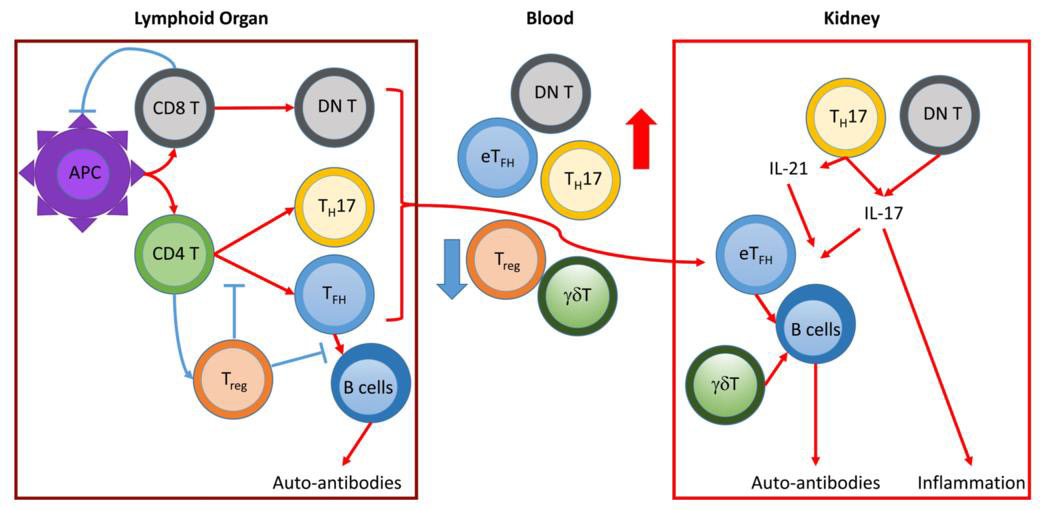
Figure 6: Roles of T cells in SLE and LN pathogenesis (red pathways up-, blue pathways downregulated) (143).
CD4+ T cell populations are generally expanded in SLE. As major contributors to autoimmunity, follicular helper T (TFH) cells proliferate significantly in SLE, providing disease-promoting B-cell targeted help for the production of autoantibodies at elevated levels [145,146]. Specifically, TFH cells promote the differentiation of B cells into memory B cells and plasma B cells, affinity maturation, and class switching. Therefore, they are causal for the increasing specificity of B cells to autoantigens and the concurrent production of autoantibodies [143,147]. Whether numbers of Interferon γ (IFNγ)-producing TH1 cells are in- or decreased in SLE has not been definitively determined and is likely related to IL-2 signaling [148,149]. TH2 cells, on the other hand, seem to rescind as mediators of SLE pathogenesis [150]. An increase in IL-17-producing CD4+ T helper (TH17) cells is accompanied by elevated serum IL-17 levels, which promotes the attraction of neutrophils and the formation of germinal centers, stimulates B cells, reduces B cell tolerance, triggers autoantibody production, and facilitates target organ damage [143,151,152]. Ultimately, CD4+ cell populations are significantly skewed towards TH17 and away from Treg cells [149].
Functional CD4+, CD8+, and γδ regulatory T (Treg) cell deficiency disrupts the physiologic balance between pro- and antiinflammatory stimuli in SLE [143]. Healthy Treg cells are known to regulate a number of processes in the immune system, among which suppression of B and T lymphocyte activation and proliferation, inhibition of autoantibody production of B cells and prevention of immunogenic but facilitating of self-tolerant DC expansion [153]. Different mechanisms have been proposed regarding the reason for deregulation of inflammatory stimuli by Treg cells: For one thing, decreased formation and survival of Treg cells has been identified, and their numbers are generally diminished. For another, defects in their immunosuppressive function are also likely [145] but might be secondary to an acquired resistance of SLE T effector cells to Treg suppression [153]. Another explanation could be the relative depletion of Treg cells, which results from activated, suppression-resistant effector T cells outweighing the number of Treg cells [151,152,154]. Lastly, the influence of TLR-dependent IL-6 expression in APCs has been shown to render effector T cells refractory to Treg suppression, a relevant mechanism given the likeliness of TLR activation by nucleic autoantigens in SLE [155].
Increased numbers of activated CD8+ T cells remarkably present with decreased cytotoxic ability, defective viral antigen responses, and therefore increased risk of infection, as well as a possible role in triggering autoimmunity [143,151]. Apart from increased numbers of functionally impaired CD8+ cytotoxic cells, quantities of NK cells are, generally, diminished (145). TCRαβ+/ CD4 /CD8- double negative (DN) T cells result from inactivation or exhaustion of autoreactive or continuously stimulated CD8+ T cells, which usually exert immunosuppressive functions in chronic infection [143]. In SLE, they have been shown to locally accumulate and contribute to infection via the production of IL-17 (156). Due to their TCRs being comprised of γ- and δ- rather than α- and β-chains, γδ T cells do not detect antigens bound to major histocompatibility complexes (MHC), but rather – similar to antibodies – bind antigens directly and exert a variety of functions [157]. By presenting antigens, producing proinflammatory cytokines, interacting with Treg cells, and promoting autoantibody production via B cell help, γδ T cells have been shown to participate in the development of SLE. Constituting around 10-15 % of T cells in the human blood under physiological conditions, their occurrence seems to be reduced in the peripheral blood but increased in affected target organs of SLE patients [158].
As opposed to thymocytes in healthy individuals, SLE T cells produce a different cytokine profile (Figure 7), which is generally characterized by an increase in IL-6, IL-17, IL-12, and IL-10, but reduced levels of TH1-dependent IFNγ and IL-2, as well as Treg-dependent transforming growth factor- β1 (TGF-β1) [149]. Notably, a decline in IL-2 is associated with impaired formation and survival of Treg cells, as well as elevated levels of proinflammatory cytokines (particularly IL-6) which inhibit Treg cell functions in SLE [151,159]. Conversely, low levels of IL-2 influence T cell subset distribution, inasmuch as skewing T cell populations towards TH17 cells, and impairing cytotoxic functions of NK and CD8+ effector T cells corresponding to SLE disease activity [148]. Also, IL-2 initiates activation-induced cell death (AICD), which is a crucial mechanism in T cells to retain peripheral autoantigen tolerance but is defective in SLE [160]. A complex set of factors influences T cell activation. Its centerpiece is the T cell receptor (TCR) or, respectively, TCR/CD3 complex, which recognizes proteolytically processed short peptide antigens bound to MHCs of both specialized APCs (predominantly MHC-II) and most nucleated cells (MHC-I). In order to be activated rather than to be put in a state of unresponsiveness (anergy), naïve T (TN) cells have to be stimulated by APCs through their T cell receptor/CD3 complexes (TCR/CD3), costimulatory signals provided by membrane molecules (CD86 or CD80 to CD28 in DC:TN ligation), and concomitant cytokines [161]. The latter decide the T cells’ fates, as T cell differentiation depends upon the availability and composition of the local cytokine environment [157].
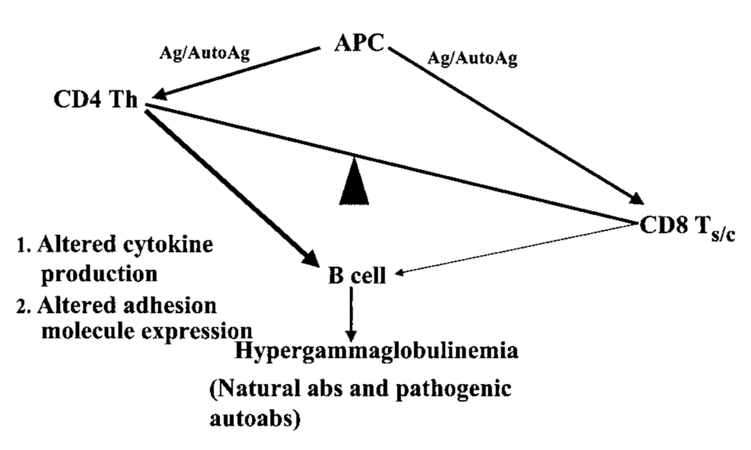
Figure 7: Disbalance of T cell function and autoantibody production (145)
SLE is associated with defects on multiple levels of the T cell signaling cascade, which can be divided into proximal, middle and distal regions ranging from extracellular ligation to protein translation. Therefore, these defects are associated with many different aberrations from premature apoptosis to dysregulated DNA methylation and impaired protein translation [145]. One of the mechanisms affecting T cell activation in a majority of SLE patients is an altered expression of Fc receptor signaling components, in particular a decrease of the T cell receptor ζ chain expressed in healthy individuals and its substitution with the more common TCR γ chain. This pathologic feature of SLE can singularly cause T cell hyperresponsiveness and diminish the TCR/CD3-mediated production of IL-2 in SLE T cells [162]. Compared to controls, hypomethylated CG-rich DNA sequences and promoters of IFNregulated genes were identified in SLE CD4+ precursor T (TH0) cells, priming thymocytes to overexpress interferon-related genes in a rapid type I-IFN response. On the other hand, hypermethylated genes like CD247, which encodes for the TCR ζ chain, have also been identified to participate in SLE T cell dysfunction [163]. As a result, commonly subthreshold interactions between T cells and APCs cause T cell activation in SLE.
Processes responsible for autoantibody production in SLE mainly proceed in secondary lymphatic tissue (Figure 8). As a general principle, activated APCs migrate from peripheral tissues to draining lymph nodes via the lymphatic system (“homing”), where they are sampled by T and B lymphocytes [164]. When a T cell is stimulated by a suitable peptide presented via a MHC, costimulatory molecules and cytokines, it differentiates into one of the many subtypes depending on the local cytokine environment [157]. If it becomes a CD4+ TFH cell, it perpetuates germinal centers by providing survival signals for activated autoreactive B cells (i.e. Centro blasts) together with follicular dendritic cells (fDC), which present autoantigens. In the subsequent process of affinity maturation, B cells are rewarded for increased specificity of their B cell receptor (BCR) to the respective autoantigen by stimulation, facilitating the expression of highly specific autoreactive SLE B cells in a proliferative process [165]. This clonal selection is based on somatic hypermutation, in which BCR/immunoglobulin (Ig) expression is genetically diversified [166]. Long-term exposure to the aforementioned survival signals causes differentiation of autoreactive B cells into memory B or class-switched, long-lived plasma B cells, which produce high levels of pathognomonic IgG autoantibodies [142]. These cells home to protective niches in the bone marrow, where they are maintained by chemokines and products of bone marrow stromal cells. This is of particular relevance in SLE pharmacotherapy, as such cells are refractory to certain immunosuppressive B cell depletion therapies [167].
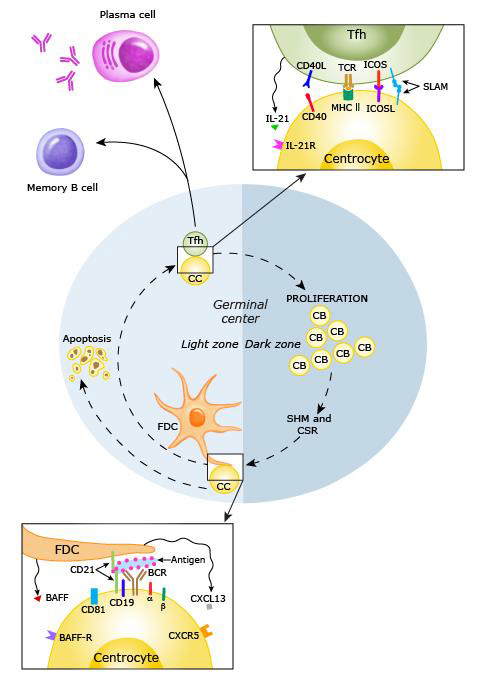
Figure 8: B cell activation in a secondary lymphoid follicle (168)
CD40 ligand (CD40L, or CD154), a surface molecule of the TNF family and TCR costimulant on (mostly CD4+) T cells, is expressed and maintained for longer durations after T cell activation in SLE [169]. As it binds to CD40 expressed on the surface of APCs, CD40L is classically known as an essential part in the co-stimulation of MHC-II antigen-presenting B cells in the process of affinity maturation, differentiation, and class switching. Hence, these mechanisms are pathologically augmented [170] in SLE T:B cell interaction. A therefore alleviated co-stimulation through T cells encounters more susceptible, hyperreactive SLE B cell phenotypes, which further promote disease activity [171]. In addition to their roles in cells of the innate immunity, TLR7 and TLR9 also activate autoreactive B cells in cytoplasmatic compartments (Figure 4.8) which internalize nucleic autoantigens or DNA/RNA-containing immune complexes, either coupled with a BCR Ig receptor [172]. TLR7 acts synergistically with CD40L and other factors in terminal B cell differentiation [173], and might, remarkably, even facilitate a thymocyte-independent mechanism of B cell activation and differentiation [174]. Additionally, TLR7 is an indispensable B-cell intrinsic mediator in the spontaneous formation of new germinal centers which induce systemic autoimmunity [175]. Nonetheless, the contribution of TLR9 in the production of autoantibodies might be ambivalent, as it competes with TLR7 for the intracellular ligand UNC93B1. For when the latter is modified for loss of TLR9 affinity, lethal TLR7-mediated inflammatory disease develops (Figure 9). Autoreactive B cells can be tied to two different sources. The first wave of B cells displaying self-reactive BCRs emanates directly from the early B-cell-repertoire in the bone marrow. Typically, these cells are deleted before exiting the bone marrow or undergo receptor editing [165].
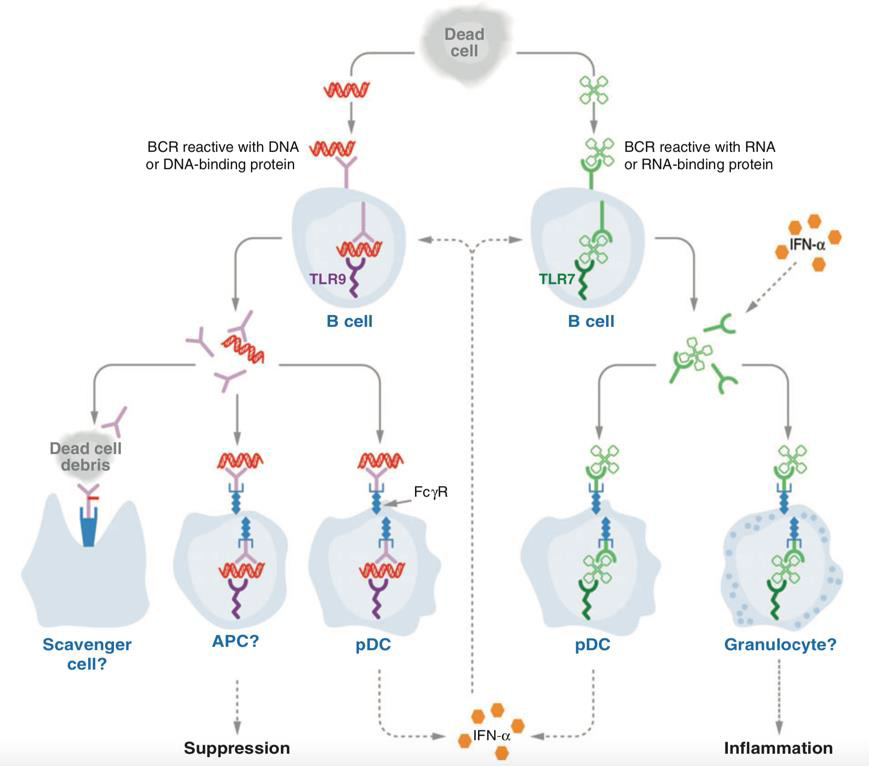
Figure 9: Roles of TLR7 and 9 in B cells (172)
These processes are attributable to central B cell tolerance checkpoints, which can be defect in SLE, resulting in a substantial primary distribution of autoreactive, but otherwise naïve B cell populations [176]. A second wave of autoreactive B cells develops in the process of somatic hypermutation of Ig (BCR) heavy and light chain variable regions. These cells are usually subject to peripheral censoring, which can be impaired by abnormalities in different B cell signaling molecules such as PTPN22, Bruton’s Tyrosine Kinase (Btk), Lyn, or B-cell activating factor (BAFF) [165]. The inhibition of mature, autoreactive B cells also relies on the surface Fc receptor FcγRIIB (CD32b), which is usually upregulated in healthy B and plasma B cells but downregulated in SLE. In normal quantities, it initiates a feedback loop upon binding IgG immune complexes, resulting in antibody homeostasis by regulating naïve B cell progression and therefore limiting the anti-DNA response to the expression of nonpathogenic IgM autoantibodies [177].
Apart from follicular responses, extrafollicular, hyperresponsive B-cell populations shape disease expression in SLE [175]. Initiating the production of somatically mutated and class switched SLE autoantibodies by long-lived plasma cells, extrafollicular B cell responses are a major contributor to circulating antibodies in SLE [143,178]. In contrast to follicular B cells, extrafollicular B-1 and marginal zone B cells generate short-lived circulating plasmablasts and extrafollicular plasma cells, which are suspected to be a main source of dsDNA autoantibodies in disease flares [165,179]. Emanating from ectopic germinal centers and lymphoid aggregates, follicular, long-lived plasma cells might also contribute to persistent autoantibody production outside of secondary lymphatic tissue [165]. Both the occurrence of circulating plasmablasts and antidsDNA antibodies is associated with an expanded population of circulating programmed cell death protein 1 (PD-1)-expressing TFH-like cells in active SLE, which are therefore likely to provide T cell help in both follicular and extrafollicular B cell maturation [143,180].
B cells have been proven to exert functions beyond autoantibody production and the buildup of B cell memory in the pathogenesis of SLE (Figure 10). For one thing, they appear to be vital APCs for the activation and priming of native T cells. For another, they promote autoimmunity of T cells reciprocally via co-stimulation of thymocytes, namely by providing CD80 and CD86 as binding partners of CD28, and CD40 to ligate CD40L. Third, they contribute to autoimmunity by proinflammatory stimulation of a wide range of targets through the excretion a number of cytokines, among which IFNα and IL- 6 [141,181].
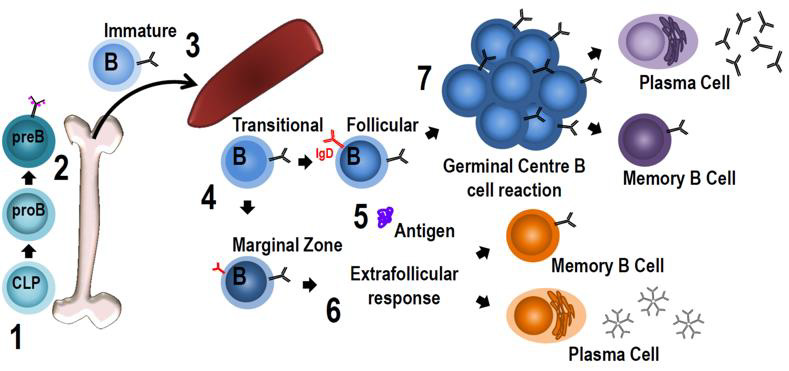
Figure 10: Stages of B cell development (147)
Autoantibodies
Pathogenic processes in SLE rely heavily on autoantibodies targeting self-antigen. Lupus-specific antibodies can be categorized according to their targets, among which DNA and DNA-binding proteins, RNA and RNA-associated proteins, β2-glycoprotein 1, phospholipids and cell membrane proteins. As an example, for pathognomonic Igs, antinuclear antibodies can be detected in serologic samples from virtually all SLE patients [182]. A number of around 180 possibly pathogenic autoantibodies have been identified, which are diverse in their prevalence and effect on disease activity but are mostly not uniquely specific for SLE. This has introduced the question whether SLE might not be a single disease with varied phenotypes but rather a similar phenotype associated with different pathogenic mechanisms. However, some 90 mutual antibodies have been shown in 20 % or more of SLE patients, and around 20 antibodies are common to more than 50 % of SLE patients [183]. Among these, anti-dsDNA and anti-Sm are most specific for SLE [1]. When comparing quantities of circulating autoantibodies, anti-Ro, anti-La, and anti-nucleosome antibodies also stand out [95]. This begs the question of commonalities among “classic” lupus autoantibodies, and indeed, nucleic acid associated antigens seem to be their preferred target. Once again, this is explained by BCR co-stimulation through T-cell derived signals, cytokines, and especially TLR7 [165].
With respect to disease patterns, certain subsets of SLE can be distinguished by related autoantibody clusters [184] or individual autoantibodies [185]. As an example, titers of 13 antibodies have been found to be significantly increased in pSLE cases with proliferative LN, among which dsDNA and C1q autoantibodies. In the same study of autoantigen microarray results, fifty autoantibodies were significantly increased in the sera of pSLE patients, including anti-BAFF, which has curiously been demonstrated to be associated with active disease [186]. Blood samples of SLE patients usually contain elevated levels of IgM, IgG and IgA autoantibodies [187]. This can be explained by the fact that the pathogenic autoantibody profile in SLE undergoes a shift from monoclonal IgM to IgG, temporally corresponding to disease progression and tissue damage (1,142). It depends on plasma cells originating from B cells undergoing affinity maturation and class switch, processes mostly driven by CD4+ TFH cells, TLRs and co-stimulation from receptors and cytokines such as IL-21 and BAFF (see above). To some extent, IgM antibodies are assumed to be a protective factor in SLE, e.g. by binding autoantigens and therefore blocking the formation of pathogenic IgG immune complexes [188]. They are also suspected to decrease IgG autoantibody production by autoreactive B cells, diminish DC activation and act as competitive inhibitors to their IgG counterparts in binding circulating nuclear antigens [187]. In autoimmune arthritis, IgM NAbs have been documented to exert a number of functions to prevent excessive, detrimental autoimmune response: T15-NAbs specific to phosphorylcholine epitopes facilitate the deposition of MBL and C1q onto apoptotic cells, enhancing their phagocytosis, and suppress TLR-induced maturation of conventional DCs. Additionally, T15-NAbs inhibit macrophage and DC secretion of pro-inflammatory cyto- and chemokines. Lastly, in a murine model, infusion of high doses of T15-NAbs or IgM anti-phosphorylcholine (PC)-inducing apoptotic cells was shown to inhibit the development of autoimmune collagen-induced arthritis [189].
In SLE, pathogenic autoantibodies contribute to disease progression in numerous ways, primarily by forming immune complexes which, as in a feedback mechanism, stimulate cells of the innate and adaptive immune system. Apart from their already discussed, essentially “upstream” immunogenic roles, autoantibodies also take part in various mechanisms of tissue inflammation, hence facilitating organ damage. Along with preformed immune complexes, which are deposited in target organs such as skin tissue or renal glomeruli, immune complexes can also be formed in situ by circulating autoantibodies binding to resident epitopes [142]. Autoantibodies generally bind (neo-)autoantigens in cellular debris exposed by NETosis, (aberrant) apoptosis or necrosis [190]. Additionally, immune complexes consisting of mammalian nucleic acids and autoantibodies are potent selfantigens for endosomal TLR7 and TLR9 activation in pDCs and therefore inductors for the excessive expression of IFNα in SLE [114]. When considering the high affinity of TLR7 to bind RNA, the fact that autoantibodies specific for RNA-binding proteins (anti- Ro, anti-La, anti-Sm) are strongly associated with high expression levels of IFN-inducible genes in PBMCs seems reasonable [120]. Consistent with the foregoing, autoantibodies against RNAcontaining antigen are more potent in the induction of type I IFN than such against DNA-containing antigen, which again underlines the proinflammatory predominance of TLR7 over TLR9 in SLE [118].
Different persistence intervals of different autoantibody classes imply different sources: While high-levels of anti-Sm, anti-Ro and anti-La antibodies are consistent with production by long-lived germinal center-derived plasma and memory B cells, transitory presence of anti-DNA antibodies suggests their production by rather short-lived, extrafollicular plasma cells [165].
Organ Damage
In SLE, tissue damage and the accompanying clinical symptoms are consequences of dysregulated autoimmune inflammatory processes and exaggerated or aberrant repair responses by resident tissue cells [191]. Tissue damage cannot be explained exhaustively only by local deposition or formation of immune complexes but depends on additional pathomechanisms of immune effector cells. This can be outlined by reference to the most prominent example of organ damage in SLE, lupus nephritis. LN is explained by deposition of immune complexes in the glomerular and tubulointerstitial compartments, the recruitment of myeloid cells and the release of enzymes from neutrophil granules and reactive oxygen intermediates from macrophages [192,193]. Recently, new concepts like the notion of immune complexes formed in situ or through NETosis (Figure 11) and the direct uptake of antibodies in different glomerular and proximal tubular cells [194] have evolved the understanding of lupus nephritis [93]. In situ, cells of the innate immune system, notably macrophages and dendritic cells, as well as certain localized cells, like glomerular endothelial and mesangial cells, produce large amounts of IFNα and IFNβ and proinflammatory cytokines via a TLR-mediated pathway. This mechanism is believed to contribute to renal damage in LN and the formation of tubuloreticular structures or inclusions [93].
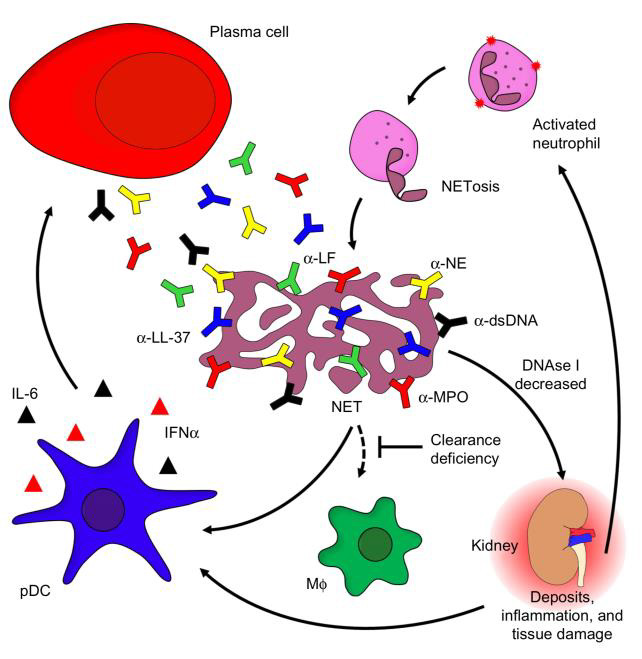
Figure 11: The role of neutrophils in SLE and target organ manifestation (142)
Exposure of extracellular DNA and chromatin fragments associated to glomerular basement membranes precedes severe nephritis and involves downregulation of DNase1 transcription and loss of nuclease activity, which might suggest a pivotal protective role for these enzymes [192]. Regarding opsonization and removal of autoantigens in the extracellular space, a protective role of the C1q has already been discussed earlier, and in the absence of C1q autoantibodies, LN is almost never seen [136]. With regard to SLE target organ damage, however, the complement system shows its Janus-faced nature: Complement factors (i.e. complement component 3 and beyond) have been demonstrated to directly cause immune-complex related renal inflammation and immunopathology [195,196]. Murine models indicate, however, that by immune complex removal, ameliorative effects of the complement system prevail and thus reduce Fcγ-receptor III (FcγRIII)-mediated inflammatory response [94]. As an additional mechanism, even antibody-deficient mice have been demonstrated to develop SLE, and pathogenic effects of B cells beyond antibody production have been identified. Namely, B cells spontaneously activate and co-stimulate pathogenic T cells, and produce a plethora of immune response-inducing cytokines. Among others, TNFα, IFNα and IL-6 mediate local inflammation following paracrine secretion. These cytokines exert a number of functions, either stimulating tissue damage by driving inflammation in an unspecific manner, by polarizing and activating T cells or by affecting lymphoid tissue formation. On the other side of the spectrum, a role for IL- 10 producing regulatory B (Breg) cells might be established, as their product, IL-10, suppresses local inflammatory processes by inhibiting further cytokine production, antigen presentation by macrophages, or TH1 and TH2 polarization [141].
Together, the ligation of TLRs, of complement receptors and fragment crystallizable receptors (FcRs) induce renal cells to express cell adhesion molecules inside the microvasculature and to release proinflammatory cyto- and chemokines [93]. Among these are type I IFNs, IFNγ, IL-6, IL-12, IL-21 and IL-23, which alter the function of local tissue (e.g., endothelial, stromal) cells and activate pathogenic lymphocytes, Mφ, and DCs on-site [1]. Due to the local expression of a limited number of chemokines, various leukocyte (sub) populations are directed into different renal compartments and locally activated [197]. Kidney tissue is foremost infiltrated by cytotoxic CD8+ T cells, TH17 cells and B cells [93]. In proximity to T cell aggregates, B cells form de novo perivascular tertiary lymphoid organs inside the kidney through clonal expansion, selection, and somatic hypermutation, namely T:B cell aggregates or germinal centers, the latter also comprising of antigen-presenting fDC cells [198]. Apart from local inflammation and tissue pathology, these aggregates contribute to intrarenal and systemic autoantibody production [199]. Certain T cell species have been shown to be particularly involved in LN pathology. IL-17 producing, CD3+/ CD4 /CD8- DN and CD3+/CD4+ T cells are generally increased in SLE patients, while TH17 and Treg cells seem to be in homeostatic balance when disease activity remains stable. In the kidney, IL 17 overexpression increases the influx of effector cells, activates B cells, increases autoantibody production and accelerates the formation of germinal centers through induction and promotion of many pathogenic mediators [200]. Additionally, activated Mo and Mφ infiltrate kidney tissue and vigorously participate in renal inflammation and injury [106]. Among Mφ subtypes, it is likely that M1 cells contribute to local inflammatory processes, but the contribution of M2 cells is less clear, as their role in resolving inflammation and tissue repair might “inadvertently” contribute to pathogenesis by proliferation and excess tissue remodeling [201].
It is obvious that LN has been a primary subject of research on target organ damage in SLE. While many different pathways for renal injury have been established, the SLE phenotype develops due to many different factors collaborating in a severely dysregulated immune response. Moreover, varying microenvironments in different tissues are believed to affect the individual contributions of participating cells and their products, e.g. interactions between myeloid cells and T cells [1]. In concert with specialized immune cells (e.g. lymphocytes, Mo/Mφ and APCs), local stromal, parenchymal and endothelial cells have been demonstrated to collaborate in target organ damage [93].
Mechanisms of Atherosclerosis in SLE
Cardiovascular disease is a common phenomenon in many autoimmune rheumatic diseases. Apart from the usual traditional and non-traditional risk factors, the vasculature is affected by harmful autoimmunity-associated, either local or systemic inflammation. Both of these inflammatory processes exhibit proatherogenic effects, promoting the formation of atherosclerotic lesions which progress and destabilize in the course of autoinflammation. Subsequently, atherosclerosis is responsible for conditions such as vessel occlusion, thromboembolism and distension or division of the vessel wall, all of which possible mediators of fatal ischemia. Atherosclerosis is the long-term result of endothelial inability to maintain homeostasis, leading to atherogenesis, a process responsible for the subendothelial accumulation of lipids and fibrous elements in large arteries resulting in a proinflammatory and procoagulant state spanning across all layers of the arterial wall [202,203]. From an inflammatory perspective, atherogenesis can roughly be divided into three stages: leukocyte adhesion, migration and activation. In a first stage, leukocytes adhere to the endothelium after the expression of different adhesion, rolling, and attachment molecules, such as vascular cell adhesion molecule-1 (VCAM- 1), P- and E-selectin, and integrins, which are expressed due to opsonization by anti-endothelial cell (EC) antibodies, inflammation, or either oxidative or mechanical stress [202,204,205].
Inflammation is suspected to be triggered by the accumulation of modified lipoprotein molecules in the arterial intima as a result of altered endothelial permeability. Early local inflammation causes the expression of pro-inflammatory cytokines such as IL-1β and TNFα, mediating endothelial expression of VCAM-1 via a NF-κB dependent pathway. Chemoattractants are of vital significance to trigger the second stage, specifically diapedesis of leukocytes entering the intima at EC junctions. Such chemokines include monocyte chemotactic protein-1 (MCP-1), Eotaxin, and chemokines induced by IFNγ (e.g. IP 10, Mig, I TAC), which attract monocytes, mast cells, and lymphocytes, respectively [202]. The third phase (Figure 12) is characterized by leukocyte activation, distinctively monocyte migration, differentiation into residual macrophages, and subsequent progression towards inflammation-perpetuating foam cells. Phagocytes are then joined by other leukocyte populations of mainly T cells, but also B cells, neutrophils, dendritic and mast cells, which locally release a multitude of, among others, pro-inflammatory, vasoactive, proteindegrading, pro- and antithrombotic substances [203,204,206]. In a continuous process, this most primitive form of atherosclerosis, or “fatty streak” develops into an inflammation-dependent, more sophisticated and enlarged lesion. In this stage, further progression into pathologically variable types of atherosclerotic plaques looms, which are generally vulnerable to progress, disrupt, and eventually cause thrombosis, embolism, or occlusion. [27,204].
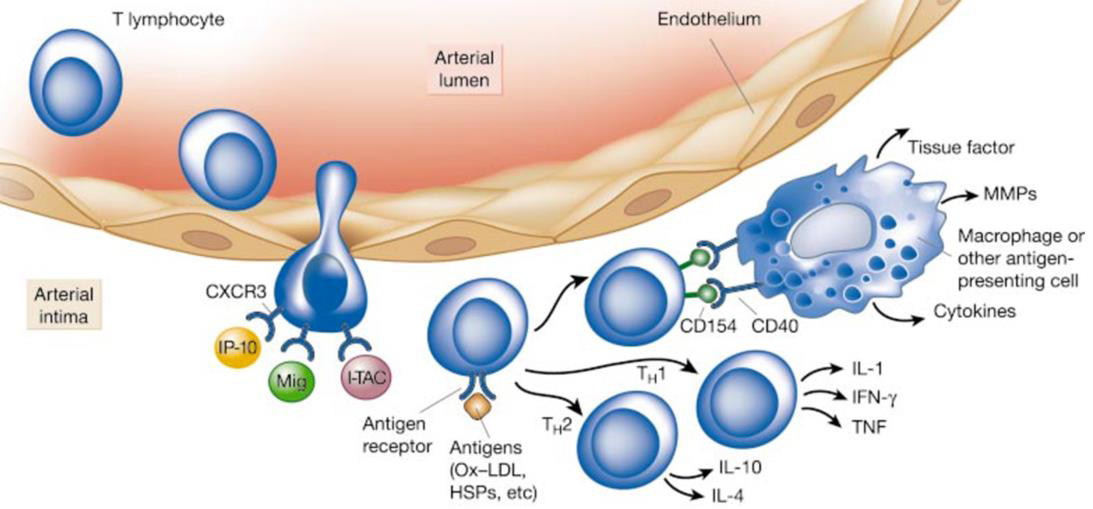
Figure 12: Inflammation in the third phase of atherogenesis (204).
When looking at these most basic processes in plaque formation, progression and rupture, initial anchor points can be discerned tying SLE to atherosclerosis. Endothelial cell necrosis, which might result from elevated levels of inflammatory mediators in the vicinity or cellular cytotoxicity [204], could be facilitated by autoreactive immune cells. This however, results in a typical henand- egg problem: As defective clearance of apoptotic and necrotic cells is present both in SLE and in atherosclerosis [27], antigen for the deposition of autoantibodies or self-reactive immune complexes is readily available. By enhancing autoinflammatory, SLE-associated processes, which result in the upregulation of the local cytokine environments, the process fueled even further. Other peculiarities of SLE, like the increased co-stimulation and recruitment of mononuclear cells via CD40L on TH cells, on which its expression is pathologically elevated, might be a source of autoimmune contribution to atherosclerosis. Indeed, soluble CD40L is increased in both SLE patients and patients suffering from acute coronary syndrome, suggesting its contribution to plaque instability and thrombosis in the setting of atherosclerosis [207]. Above and beyond the mentioned pathways, SLE contributes to atherosclerosis in a number of ways through unspecific upregulation of the immune system, e.g., through cytokine responses, up- and downregulation of the complement pathway or inflammation-associated prothrombotic disposition [202]. Besides the atherogenic involvement of the SLE-governed immune system, its contribution in anti-atherogenic processes is also feasible. In this respect, induced natural killer T (iNKT) cells have been identified as possible mediators of early stage, but not clinical apparent atherosclerosis in SLE.
Acting as disease-ameliorating effectors by responding to lipid antigen presentation in APCs, iNKT cells have been shown to release a wide array of anti-inflammatory cytokines and to polarize macrophages into an anti-inflammatory and anti-atherogenic M2 phenotype [208]. It is, however, not clear if the apparent reduction and dysfunction of iNKT cells in SLE patients impairs a possible, more extensive atheroprotective role [209]. As one of the major tissues affected by SLE pathology, the endothelium is a susceptible target for autoinflammation. It exerts pivotal functions of immunity: ECs express PRRs, produce IL-1 and other inflammatory molecules, internalize ligands such as low-density lipoprotein (LDL) particles, present antigens to specific T cells, recruit leukocytes, increase permeability, and cause edema (210). The endothelium happens to also be extensively involved in atherosclerosis, which is accompanied by loss or disturbance of its various regulatory functions [204,211]. Nitric oxide (NO), a vasodilator whose reduction is associated with endothelial dysfunction, reduces the expression of VCAM-1 by inhibition of NF-κB, the central transcriptional control point in vascular inflammation [204]. Decreased bioavailability of NO links inflammation to endothelial dysfunction, and is caused by elevated levels of TNFα, which downregulates the expression of endothelial NO synthase (eNOS). Reactive oxygen species (ROS), mediated by inflammatory cytokines, further reduce the availability of NO in the vessel wall, therefore causing impaired endothelium-dependent vasorelaxation and increased arterial stiffness [27,84]. Endothelial dysfunction is characterized by an increase in permeability, loss of junctions, secondary apoptosis, and therefore leukocyte diapedesis [205]. Resulting from direct binding of autoantibodies to endothelial cells or from the deposition of pre-formed immune complexes and subsequent autoinflammatory responses, it primarily causes functional impairment and prothrombotic activity of the arterial wall (Figure 13). Therefore, endothelial dysfunction makes for a proinflammatory and proadhesive entry point for subsequent atherosclerotic transformation, linking it to local autoinflammation [211].
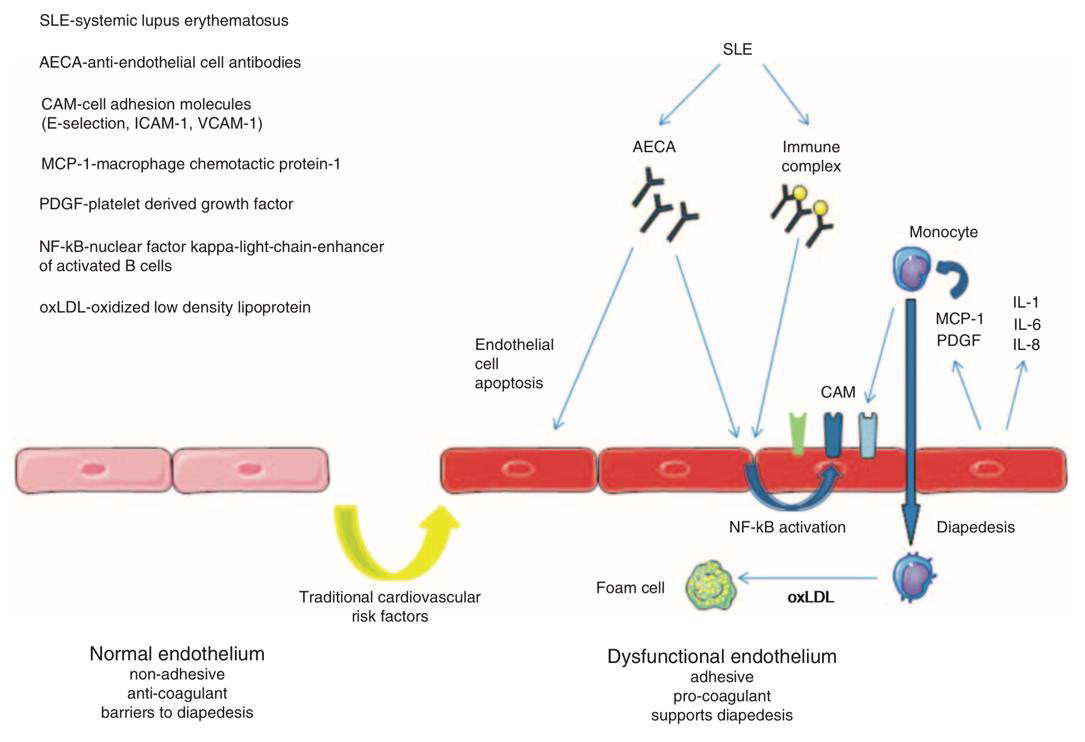
Figure 13: Autoantibodies causing endothelial injury in SLE (205).
A number of processes facilitate the migration of Mo, the uptake of LDL into Mφ and therefore the development of foam cells. On the other hand, the metabolism of triglycerides (TG) and LDL as well as the efflux of cholesterol are impaired in SLE through a decrease of lipoprotein lipase and cholesterol 27-hydroxylase. When exposed to ROS, LDL is converted to oxidized LDL (oxLDL), which stimulates the influx of Mo and the subsequent differentiation into Mφ via MCP- 1 and macrophage colony-stimulating factor (M-CSF). Therefore, oxLDL subsequently leads to its own continued phagocytosis via scavenger receptor CD36, but inhibits phagocytosis of apoptotic cells, possibly further contributing to autoinflammation in SLE [202]. Moreover, in-situ formation and deposition of oxLDLcontaining immune complexes are likely to occur as such complexes have been identified in human atherosclerotic lesions, further driving autoimmunity and -inflammation [212]. Apart from exposure to oxidative stress, increased oxLDL levels can be attributed to several other mechanisms, such as anti-highdensity lipoprotein (HDL), anti-Apolipoprotein A1 (ApoA1) and antiphospholipid autoantibody-mediated, reduced levels of antioxidant HDL and reduced paraoxonase activity in HDL particles [4,208,213]. Among the reasons for elevated levels of oxLDL is also an increase in detrimental pro-inflammatory HDL (piHDL), which occurs during states of systemic inflammation and oxidative stress, and is, conversely, strongly associated with atherosclerosis in SLE, likely contributing to the condition [213-216].
Complement activation might also contribute to the process of endothelial cell activation and recruitment of leukocytes to inflammatory sites [205]. In SLE-associated atherosclerosis, it has been shown to precede the development of spontaneous atherosclerotic lesions [210]. A protective or pathogenic role of C1q in SLE has not yet been fully established [205], but C3, C5 and overall complement activity has been demonstrated to correlate with subclinical markers of atherosclerosis, therefore reflecting activation of the complement system in patients with atherosclerosis and SLE. Interestingly, piHDL particles have been directly correlated to complement levels, suggesting an additional influence of C3 and C4-enriched HDL. Also, the complement membrane attack complex has been demonstrated to correlate to plaque formation, a finding consistent with various traces of complement activation in atherosclerotic lesions in the general population [217]. Considering the above, a contribution of the classical complement pathway in SLE-associated atherosclerosis seems likely, bearing in mind its dependence on antibodies, which have been proven to be associated with both conditions (see below). Additionally, complement induces the release of, among other molecules, MCP-1 from human VSMCs, which are – under physiological conditions – well protected from the exposure to complement by the tunica intima, but become exposed to plasma molecules in inflammation [218].
Among innate immune cells contributing to atherosclerosis, a special role for neutrophils has become apparent, with the main stage being taken by LDGs, a distinct subset of neutrophils in SLE patients with the ability to initiate different atherogenic processes [132]. Among these, LDGs propagate apoptosis, dysfunctional endothelial repair, atherothrombosis, and NET formation [27]. With regard to the latter, neutrophil extracellular traps contain antimicrobial molecules which induce endothelial cell death and vascular dysfunction [132,202]. NETosis-prone LDGs further contribute to accelerated atherosclerosis by disrupting the maturation of endothelial progenitor cells (EPC), by directly inducing endothelial damage or synthesizing increased levels of pro-inflammatory cytokines (IFNα, IL-17) (4,202). Furthermore, NETs drive IFN, IL-1β and IL-17 expression in target myeloid cells, which promote a vicious cycle of tissue inflammation [27]. Moreover, oxidant-generating enzymes in NETs lead to the oxidation of HDL and puts it into a dysfunctional, pro-inflammatory state [213,215,219]. Considering the fact that oxLDL has the ability to mediate NET release [220], another vicious cycle might be driven by dysfunctional removal of oxLDL as a consequence of HDL oxidation, further propagating LDGs to form NETs. Taking into consideration the impaired clearance of cell remnants in SLE, and degradation protective anti-DNAse I and anti-NET autoantibodies, NETs are well protected against cellular clearance [202]. This, in turn, stimulates autoinflammation through the exposure of potent autoantigens such as dsDNA and various SLE-dependent immune complexes [132].
In conjunction with peculiarities of SLE, a generally proinflammatory, self-perpetuating feedback mechanism of NETs can be discerned in atherosclerosis, and unsurprisingly, NETs remain present in mature atherosclerotic plaques, further implying their role in atherosclerotic progression [202]. Decreased numbers and decreased capacity of EPCs and myeloid circulating angiogenic cells (MACs) have been described in SLE patients with atherosclerosis. Hence, their capacity to mediate angiogenesis and therefore revascularization and vascular repair by migration, differentiation and production of growth factors, predominantly vascular endothelial growth factor (VEGF) and hepatic growth factor, is impaired [221-223]. Apart from type I IFNs, lowered vitamin D levels have been associated with these findings [4,222,224]. Vitamin D deficiency has been described as a likely proatherogenic factor in aSLE and pSLE patients, but evidence has so far been inconclusive [87,225]. Even so, vitamin D has been shown to positively modulate endothelial function in SLE patients, whereas in vitro, MAC function has been augmented by calcitriol [226]. As pathognomonic mediators of SLE, type I IFNs have been suggested as major contributors to atherogenesis. They promote foam cell formation, platelet activation and an imbalance between endothelial damage and repair (4,27). By inducing apoptosis of EC, vascular rarefication is promoted.
It is joined by dysfunctional vasculogenesis, correlating to dysfunctional EPCs and depleted numbers thereof, as well as dysfunction of resident endothelial cells [202,222–224]. Type I IFNs recruit Mφ to atherosclerotic lesions, promoting their lipid uptake through scavenger receptors and thus stimulating foam cell formation, enhancing atherosclerotic plaques. Apart from pro-inflammatory stimuli by type I IFNs, they exert prothrombotic effects, prevent maturation of vascular smooth muscle cells (VSMCs), cause VSMC dysfunction, retardation and apoptosis, and increase the risk of cardiovascular events by rupture of destabilized plaques [4,27,128,227,228]. In addition to the stimulation of phagocytes, increased levels of IFNα in atherosclerotic plaques enhance vascular damage directly by cytotoxic T and NK cells or mediated indirectly via TH, most notably apoptosis of ECs and VSMCs via a TRAILdependent mechanism [206]. Consistent with these findings, type I IFNs have been shown to be potent antiangiogenic factors which are independently associated with different markers of subclinical atherosclerosis in SLE patients [128,206,223]. As strong promoters of type I IFN production by pDCs (see above), TLR7 and TLR9 might therefore play an important role in the formation and progression of atherosclerotic lesions [206]. Apart from type I IFN, apoptotic EC microparticles also cause expression of pro-inflammatory cytokines such as IL-6 and TNFα by pDCs and mDCs, which demonstrates another possible self-perpetuating mechanism of atherosclerotic lesions [229].
One of the most overexpressed non-type I IFN cytokines in SLE, IFNγ, contributes to plaque formation by upregulating a number of atherogenic processes, among which antigen presentation or cytokine (TNFα, IL-1) expression [202]. It also mediates the inhibition of collagen production in VSMC and the overexpression of collagenases (Matrix Metalloproteinases, or MMP 1, 8, 13) and gelatinases (MMP 2, 9) in mononuclear phagocytes, endothelial and smooth muscle cells via IL-1β, TNFα and CD40L (204). Therefore, IFNγ is a powerful growth inhibitor for VSMC and EC in the local environment that promotes instability of atherosclerotic plaques [230]. In concert with TNFα, IL-1 induces local inflammation by stimulation of Mφ, induces MMPs, and promotes the production of cell surface adhesion molecules and colony stimulation factors (G-, M-, GM-CSF). TNFα has been found in all stages of atherosclerotic lesions, but may not be specifically etiological in rheumatic disease, as it also presents in atherosclerosis among the general population [230]. Other cytokines might also promote or ameliorate the pathogenesis of SLE. Among these, decreased levels of transforming growth factor β (TGF-β) were associated with arterial wall dysfunction and premature atherosclerosis [202]. More precisely, oxLDL inhibits the activation of latent TGF-β1 and (V)LDL inhibits the binding of TGF-β1 to the type II TGF-β receptor, thereby suppressing signaling. Low levels of this cytokine are strongly associated with increased CIMT, likely resulting from EC and VSMC proliferation accompanied by excessive apoptosis and atherosclerotic disposition [231].
So far, atherogenic processes have been outlined as mainly innate immunity-dependent, but adaptive immunity also plays a major role in atherosclerosis. In addition to circulatory lymphocytes, adventitial artery tertiary lymphoid organs (ATLOs), a form of tertiary lymphatic tissue, develop as a result of chronic local inflammation and contribute to atherosclerosis in SLE locally [203]. ATLOs persist in the connective tissue surrounding atherosclerotic arteries and can range from small T/B cell clusters to lymph nodelike organs harboring lymphocytes and various effector cells of innate immunity. As they can be hugely complex, it has not been fully understood whether ATLOs exert disease-promoting or -ameliorating functions, or both [232].
Atherosclerosis is likely caused by an imbalance between pathogenic T cells and Treg cells but establishing clinical manifestations of this phenomenon has been a challenging task in SLE patients to date [233]. Nevertheless, a disbalance of TH17 and Treg subgroups in patients with atherosclerosis and SLE has been suggested [234], implicating a specific role for a IL-17 mediated response in atherogenesis [212]. Conversely, Treg cells may protect against atherosclerosis by improving endothelial function and inhibiting B cell activation and the production of inflammatory cytokines [27]. Complicating the possible contribution of TH17 cells, it might even reduce vascular T cell infiltration and development of atherosclerosis [144], leaving the question of its precise role unanswered.
Different mechanisms have been described for a potential of SLE B cells to promote atherosclerosis, although various B cell subpopulations seem to exert opposing effects. In SLE, autoantibody production of B cells generally shifts from largely atheroprotective IgM NAbs to largely pathogenic IgG autoantibodies [235]. While both follicular and extrafollicular B cells might contribute to atherosclerosis in SLE by producing class-switched IgG antibodies, follicular B cells predominate in releasing pathogenic IgG antibodies and proatherogenic cytokines [203], likely due to stimulatory effects of BAFF [236]. FcγRIIB has already been discussed as an inhibitory Fc receptor to IgG autoantibodies, that is typically downregulated in SLE (see above). As it inhibits pro-inflammatory signaling in B cells, the lack of FcγRIIB has been proven to exacerbate atherosclerosis [237], which might exactly be its role in SLE. B-cell derived autoantibodies can be directed against a variety of epitopes, e.g. endothelial cells, phospholipids and oxLDL, and traditional SLE antigens such as dsDNA. For some SLE IgG autoantibodies, a distinct pathogenic role in atherosclerosis has been assumed, but no underlying mechanism has yet been identified [238]. In about twothirds of SLE patients, anti-endothelial cell (AEC) autoantibodies are present, exposing a potential to upregulate adhesion molecule expression, induce oxidative stress, or cause apoptosis in EC [4,205,239]. Sub-APS levels of antiphospholipid autoantibodies contribute to a high cardiovascular disease risk profile [35] and may also be directly causative for atherosclerosis in SLE [4].
Anti-β2-glycoprotein I (β2-GPI)-autoantibodies, a subset of antiphospholipid antibodies, have been linked to subclinical atherosclerosis in SLE patients. They facilitate oxLDL foamcell- generating phagocytosis via an anti-β-glycoprotein I (β2- GPI)-dependent mechanism and are also intertwined with increased T cell reactivity, an increase in NETs, and thrombin production [212,240]. While anti-oxLDL antibodies have been loosely associated with atherosclerosis, anti-oxidized palmitoyl arachidonoyl phosphocholine (oxPAPC, a more stable epitope of oxLDL) antibodies do indeed correlate to Intima-Media-Thickness (IMT), and therefore to subclinical atherosclerosis [202,238]. Autoantibodies to lipoprotein lipase (LPL) impair degradation of very low-density lipoprotein (VLDL) conversion to LDL, resulting in a low LDL, low HDL, high VLDL, high TG lupus lipid pattern, which is correlated with disease activity and markers of inflammation [223]. Other populations of autoantibodies such as anti-cardiolipin and anti-dsDNA antibodies have also been demonstrated to correlate with atherosclerotic markers and events in SLE [241], but whether they contribute in an atherosclerosis-specific or general SLE pathomechanisms is yet to be understood. In contrast to pathogenic autoantibodies, the lack of distinct subsets of protective autoantibodies has been shown to be correlated with cardiovascular disease in SLE patients (Figure 14). For one thing, decreased levels of IgM NAbs to phosphorylcholine further impair apoptotic cell clearance in SLE and are associated with carotid plaque formation and increased vulnerability (4,242). Analogously, antibodymediated clearance of LDL particles might also be impaired, which is reflected in decreased serum levels of atheroprotective anti- ApoB-100 IgG and IgM NAbs [243].
Figure 14: Atheroprotective and proatherogenic roles of B cells, NAbs, and pathogenic autoantibodies (235)
As already outlined previously, data resulting from smaller samples and case studies suggest that, in addition to the wellestablished correlation between SLE and atherosclerosis, patients suffering from SLE are at an increased risk of developing arterial, mostly aortic or coronary aneurysm. Literature on the mechanism of its development in SLE is rather scarce compared to such of atherosclerosis. Formation of AA is described as an immunerelated process in which cells of the innate and adaptive immune system accumulate in the adventitia of the aortic wall [73]. Apart from this, the pathogeneses of AA and dissection have also been linked to atherosclerosis, aortic elastic tissue degeneration and certain vasculitides [72,73,244,245]. This corresponds with the fact that, seemingly, some consent has been reached about at least two different SLE-related disease mechanisms which result in the development of aortic aneurysms. The first of the mentioned pathway is one of atherosclerosis-associated, mostly abdominal AA formation. Sites of aneurysm often display atherosclerosis, which increases the likelihood to rupture plaques as a result of aneurysmatic dilatation [246]. As atherosclerosis and abdominal AA are also positively correlated among both the general population and SLE patients [56,69,72,247], this pathomechanism might be similar to such of non-SLE-related abdominal AA. This is supported by the fact that abdominal AAs are more likely to occur in hypertensive, older SLE patients [71], which might lead to the assumption that general risk factors of abdominal AA also apply to SLE patients. Therefore, abdominal AA might be fueled by secondary hypertension due to damage of renal vasculature in SLE, which has also been demonstrated as an independent risk factor for the development of aneurysm [56].
The second pathway, which is hypothesized to account for most cases of thoracic AA in SLE patients, is believed to result from a SLE-related inflammatory process that leads to a diffuse pattern of aortitis which is associated with a higher prevalence of combined aneurysm and dissection [244,245,248]. This pathway might result from vasculitis-like inflammation [56], which might be similar or even equivalent to either Takayasu’s arteritis or cystic medial degeneration on a case-to-case basis [72,249]. The fact that mostly young and normotensive patients are affected might imply a lower dependence on mechanical stress compared to atherosclerosisassociated aneurysms [71]. This “vasculitis-like” pathway and atherosclerosis might also be closely related by sharing a similar autoinflammatory process [246], which is, however, likely to extend beyond the lamina intima in the case of atherosclerosis. Evidence of aneurysms beyond of the aortic wall in SLE is extremely rare. As an example, a causative relation between SLE and coronary artery aneurysm is suggested, but its mechanism is poorly understood [69]. It might result from severe coronary inflammation, medial degeneration and weakening of the arterial wall by deposition of Igs and complement, suggesting a similar, “vasculitis-like” pathway [42,69,247].
Discussion
This review has demonstrated a substantial array of pathomechanisms contributing to SLE onset and progression. Many of these general mechanisms of disease are reflected in atherosclerotic processes and have been suspected to facilitate atherogenesis. Starting from the most exposed layer of the vessel wall, ECs contribute to autoinflammation both as active mediators and as targets for autoimmunity. Apart from the endothelium’s role as a regulator of mechanical stress and lipoprotein uptake, it also exerts immune functions and influences inflammatory processes. Unless it induces endothelial cell death, autoimmunity in SLE is likely to impair homeostasis and skew ECs towards a more pro-inflammatory, hyporegenerative state. Even though the endothelium has been a primary subject of prior research, VSMCs might also exert pro-inflammatory functions and fail to retain their physiological functionality, primarily leading to a destabilization of the vessel wall. While the reviewed literature has not specifically highlighted other resident cells as possible mediators of SLEassociated atherosclerosis, cytokine-expressing fibroblasts are only one likely, additional option of such contribution.
Possible interfaces between immune cells in SLE and atherosclerosis are versatile. First and foremost, Mo/Mφ play a dominant role in atherogenesis. While their capacity to phagocytose cellular remnants is decreased, foam cell formation is preserved, most likely due to SLE-derived costimulatory influences and the occurrence of pro-inflammatory subpopulations. A distinct subgroup of neutrophils, namely NETosis-prone LDGs, interferes with vessel wall homeostasis in a number of ways, completing their general relevance in SLE as main drivers of NET formation. DCs, T and B cell populations facilitate excessive inflammation by making their SLE-specific, overshooting contributions. Among these, the production of pathogenic autoantibodies, which may even originate from tertiary germinal centers adjacent to atherosclerotic plaques, stands out most prominently. Such autoantibodies are either fairly specific to sites of atherosclerosis, as is the case with AEC, anti-β-GP1 and anti-oxLDL, or rather unspecific. The latter may target autoantigen in areas with a high concentration thereof, such as atherosclerotic plaques. Apart from the production of autoantibodies, upregulated T cell help and excessive cytokine production are just two of many possible cellular contributions to SLE-associated atherosclerosis. Completing the role of the immune system, the complement cascade has been shown to activate in atherosclerotic lesions, suggesting a direct influence on endothelial cells and lipoproteins, as well as more unspecific effects at sites of active inflammation. Beyond its immune phenotype, SLE has been shown to promote the formation of pro-inflammatory and otherwise dysfunctional HDL, oxLDL, and prothrombotic environments, which have been shown to promote atherogenesis.
On par with its underlying disease, SLE-associated atherosclerosis has been discussed as a combination of many virtually independent mechanistic defects, but an indispensable trigger has not been identified for either condition. Because SLE presents as an exceptionally heterogenous disease, the very likely explanation would suggest a general disbalance of immunity, causing homeostasis to tip over as soon as pro-inflammatory outweigh regulatory stimuli. Considering the many possible vicious cycles in SLE, this notion is a compelling explanation for the correlation between disease activity over time and prevalence of atherosclerosis. While some SLE-specific defects have been shown to prevail in distinct cells of the immune system, others span across many systems. Apart from ubiquitous mediators such as type I IFNs, individual contributors to cell signaling reach beyond certain cell types. As such, TLR7 and TLR9 run like a common thread through many parts of autoimmunity in SLE, and therefore constitute likely candidates for further research into the vascular manifestations of SLE.
Undeniably, this review has certain limitations. Notwithstanding the vast amount of literature on the pathogenesis of SLE and SLEassociated atherosclerosis, many questions still remain unanswered. Among these, one certainly concerns the lack of research into the pathogenesis of SLE-associated aneurysms. Notwithstanding the fact that two distinct pathways of aneurysm and dissection formation in SLE have been identified, the corresponding pathomechanisms have not yet been illuminated. Considering the underlying mechanisms, autoimmune processes involving the enzymatic degradation of connective tissue might only be one of many fields of interest. In contrast to this mostly uninvestigated manifestation, independent disease entities such as large- and medium-vessel vasculitides were not reviewed in this thesis due to their self-sufficient nature. These conditions can result from SLE or share certain pathomechanisms with SLE, both of which might translate into their contribution to SLE-associated atherosclerosis or aneurysm. Lastly, as a general principle, iatrogenic interventions were not described as separate mediators of disease because the literature cannot be distinguished between data from treated vs. treatment-naïve individuals. In this regard, it is important to remark that disease-modulating or anti-inflammatory treatment regimens may damage tissue via discrete mechanisms.
Conclusion
This review of the literature discussed the main disease mechanisms in SLE known to date and many peculiarities of these mechanisms. Both longtime knowledge as well as more recent insights into the pathomechanisms of SLE were outlined, resulting in a broad overview as well as a more detailed look into certain aspects of SLE pathogenesis. Strikingly, SLE pathomechanisms could not be delineated as a single, stringent process. Rather, the pathophysiology of SLE presents as a complex and convoluted set of individual immune dysregulations, in which components of the innate and adaptive immune systems partake. Apart from its etiologic diversity, SLE results in profoundly different clinical pictures, which led to the question whether SLE is a single disease entity or a rather a collective term for different conditions. Some of its various manifestations are of vascular nature, the most prominent being atherosclerosis. Along with aneurysm and dissection, atherosclerosis remarkably affects SLE patients at much younger age than the general population and correlates with disease duration and activity. Therefore, these vascular manifestations are likely to be associated to the pathomechanisms of SLE. Indeed, many of such mechanisms have been identified to take part in accelerated atherosclerosis, but no definitive trigger can be singled out. Rather, an interplay of different autoinflammatory reactions likely facilitates many stages of atherogenesis. Although no definitive mechanisms have yet been established, two individual pathogeneses are proposed to cause SLE-associated aneurysms and dissections in different patient cohorts. While the first presumed mechanism is dependent on pre-formed atherosclerotic plaques, the second might share a common autoinflammatory pathway with SLE-associated atherosclerosis.
For More Articles: Biomedical Journal Impact Factor: https://biomedres.us
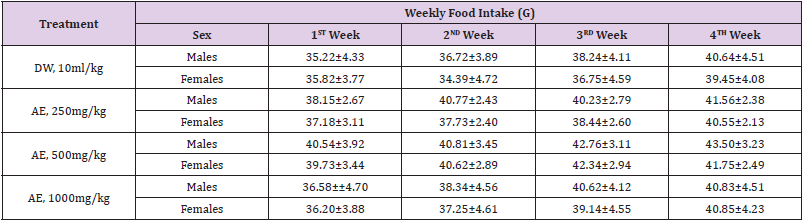
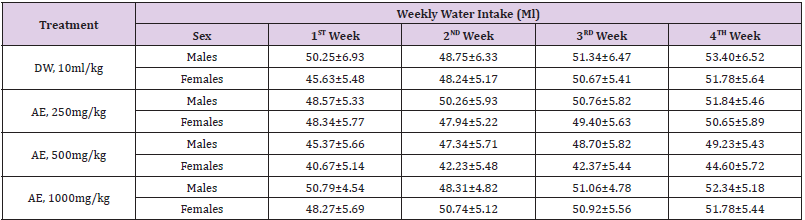
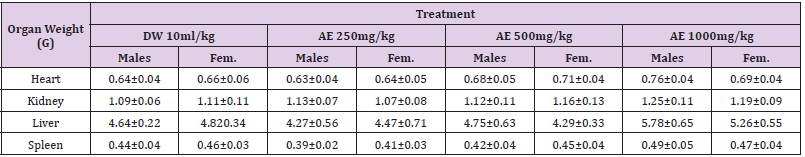
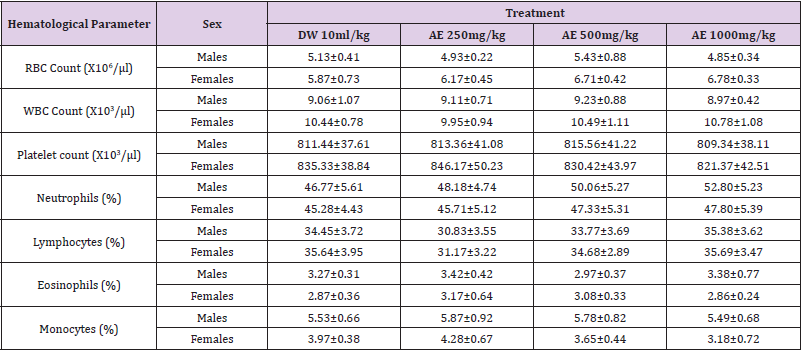
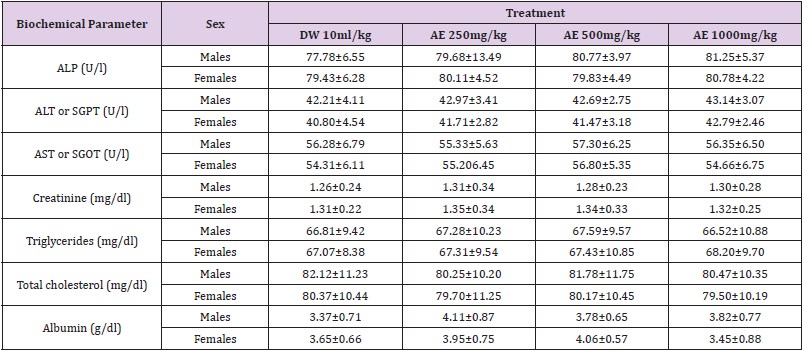
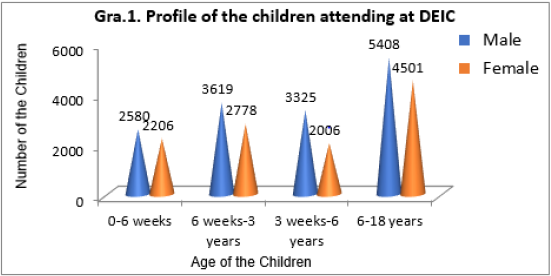
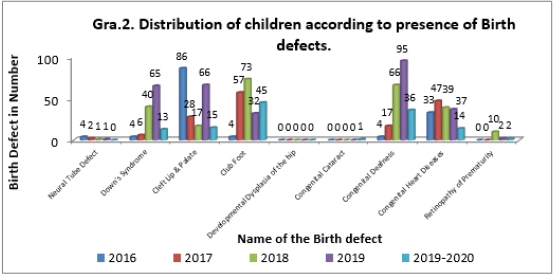
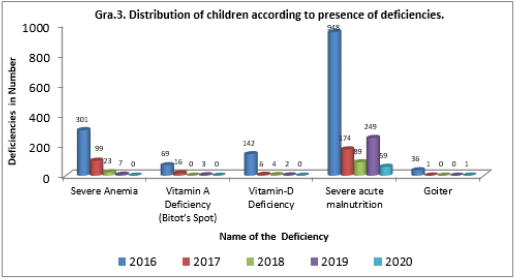
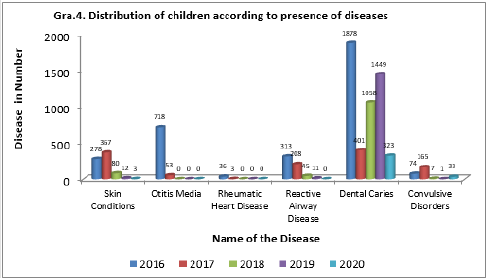
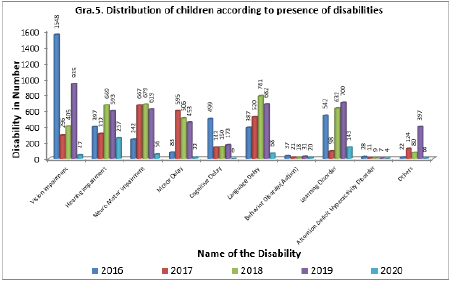

 Loss of weight in drying at 105°C : 7.09Absolute alcohol solubility : 3.20Water solubility : 21.20
Loss of weight in drying at 105°C : 7.09Absolute alcohol solubility : 3.20Water solubility : 21.20